+ Active users of the gget website since its creation: +
+ +[](https://github.com/lauraluebbert/lauraluebbert) + +# 🧑🤝🧑 Dependents +The following applications build on *gget*: +- [Therapeutics Data Commons (TDC)](https://tdcommons.ai/) + Artificial intelligence foundation for therapeutic science ([source code](https://github.com/mims-harvard/TDC), [Nat Chem Bio paper](https://www.nature.com/articles/s41589-022-01131-2)) by +Harvard's Artificial Intelligence for Medicine and Science lab. +- [BioDiscoveryAgent](https://github.com/snap-stanford/BioDiscoveryAgent) + BioDiscoveryAgent is an LLM-based AI agent for closed-loop design of genetic perturbation experiments ([preprint](https://arxiv.org/abs/2405.17631)) by the Stanford Network Analysis Project. +- [DeepChopper](https://ylab-hi.github.io/DeepChopper/) + Language models to identify chimeric artificial reads in NanoPore direct-RNA sequencing data by the Yang lab at Northwestern. +- [BRAD](https://github.com/Jpickard1/BRAD) + A LLM powered chatbot for bioinformatics ([documentation](https://brad-bioinformatics-retrieval-augmented-data.readthedocs.io/en/latest/index.html), [project main page](https://brad-bioinformatics-retrieval-augmented-data.readthedocs.io/_/downloads/en/latest/pdf/)). +- [scPRINT](https://www.jkobject.com/scPRINT/) + scPRINT is a large transformer model built for the inference of gene networks (connections between genes explaining the cell's expression profile) from scRNAseq data ([preprint](https://www.biorxiv.org/content/10.1101/2024.07.29.605556v1)). +- [AnoPrimer](https://sanjaynagi.github.io/AnoPrimer/landing-page.html) + AnoPrimer is a Python package for primer design in *An. gambiae* and *An. funestus*, whilst considering genetic variation in wild whole-genome sequenced specimens in malariagen_data. +- [AvaTaR](https://github.com/zou-group/avatar) + Optimizing LLM Agents for Tool-Assisted Knowledge Retrieval (NeurIPS 2024) by James Zou Lab at Stanford University. +- [GRLDrugProp](https://github.com/Madscba/GRLDrugProp) + Graph representation learning for modelling drug properties. +- Rust implementation of *gget*: [https://github.com/noamteyssier/ggetrs](https://github.com/noamteyssier/ggetrs) +- [https://github.com/Superbio-ai/getbio](https://github.com/Superbio-ai/getbio) +- [https://github.com/yonniejon/AchillesPrediction](https://github.com/yonniejon/AchillesPrediction) +- [https://github.com/ELELAB/cancermuts](https://github.com/ELELAB/cancermuts) +- [https://github.com/Benoitdw/SNPrimer](https://github.com/Benoitdw/SNPrimer) +- [https://github.com/louisjoecodes/a16z-hackathon-project](https://github.com/louisjoecodes/a16z-hackathon-project) +- [https://github.com/EvX57/BACE1-Drug-Discovery](https://github.com/EvX57/BACE1-Drug-Discovery) +- [https://github.com/vecerkovakaterina/hidden-genes-msc](https://github.com/vecerkovakaterina/hidden-genes-msc) +- [https://github.com/vecerkovakaterina/llm_bioinfo_agent](https://github.com/vecerkovakaterina/llm_bioinfo_agent) +- [https://github.com/greedjar74/upstage_AI_Lab](https://github.com/greedjar74/upstage_AI_Lab) +- [https://github.com/alphavector/all](https://github.com/alphavector/all) + +Also see: [https://github.com/pachterlab/gget/network/dependents](https://github.com/pachterlab/gget/network/dependents) + +# 📃 Featured publications +- David Bradley et al., [The fitness cost of spurious phosphorylation.](https://doi.org/10.1038/s44318-024-00200-7) *The EMBO Journal* (2024). DOI: 10.1038/s44318-024-00200-7 +- Mikael Nilsson et al., [Resolving thyroid lineage cell trajectories merging into a dual endocrine gland in mammals.](https://doi.org/10.21203/rs.3.rs-5278325/v1) *Nature Portfolio (under review)* (2024). DOI: 10.21203/rs.3.rs-5278325/v1 +- Avasthi P et al., [Repeat expansions associated with human disease are present in diverse organisms.](https://doi.org/10.57844/arcadia-e367-8b55) *Arcadia* (2024). DOI: 10.57844/arcadia-e367-8b55 +- Ibrahim Al Rayyes et al., [Single-Cell Transcriptomics Reveals the Molecular Logic Underlying Ca2+ Signaling Diversity in Human and Mouse Brain.](https://doi.org/10.1101/2024.04.26.591400) *bioRxiv* (2024). DOI: 10.1101/2024.04.26.591400 +- David R. Blair & Neil Risch. [Dissecting the Reduced Penetrance of Putative Loss-of-Function Variants in Population-Scale Biobanks.](https://doi.org/10.1101/2024.09.23.24314008) *medRxiv* (2024). DOI: 10.1101/2024.09.23.24314008 +- Shanmugampillai Jeyarajaguru Kabilan et al., [Molecular modelling approaches for the identification of potent Sodium-Glucose Cotransporter 2 inhibitors from Boerhavia diffusa for the potential treatment of chronic kidney disease.](https://doi.org/10.21203/rs.3.rs-4520611/v1) *Journal of Computer-Aided Molecular Design (under review)* (2024). DOI: 10.21203/rs.3.rs-4520611/v1 +- Joseph M Rich et al., [The impact of package selection and versioning on single-cell RNA-seq analysis.](https://pmc.ncbi.nlm.nih.gov/articles/PMC11014608/#:~:text=10.1101/2024.04.04.588111) *bioRxiv* (2024). DOI: 10.1101/2024.04.04.588111 +- Sanjay C. Nagi et al., [AnoPrimer: Primer Design in malaria vectors informed by range-wide genomic variation.](https://wellcomeopenresearch.org/articles/9-255/v1) *Wellcome Open Research* (2024). +- Yasmin Makki Mohialden et al., [A survey of the most recent Python packages for use in biology.](http://dx.doi.org/10.48047/NQ.2023.21.2.NQ23029) *NeuroQuantology* (2023). DOI: 10.48047/NQ.2023.21.2.NQ23029 +- Kimberly Siletti et al., [Transcriptomic diversity of cell types across the adult human brain.](https://doi.org/10.1126/science.add7046) *Science* (2023). DOI: 10.1126/science.add7046 +- Beatriz Beamud et al., [Genetic determinants of host tropism in Klebsiella phages.](https://doi.org/10.1016/j.celrep.2023.112048) *Cell Reports* (2023). DOI: 10.1016/j.celrep.2023.112048 +- Nicola A. Kearns et al., [Generation and molecular characterization of human pluripotent stem cell-derived pharyngeal foregut endoderm.](https://doi.org/10.1016/j.devcel.2023.08.024) *Cell Reports* (2023). DOI: 10.1016/j.devcel.2023.08.024 +- Jonathan Rosenski et al., [Predicting gene knockout effects from expression data.](https://link.springer.com/article/10.1186/s12920-023-01446-6) *BMC Medical Genomics* (2023). DOI: 10.1186/s12920-023-01446-6 +- Peter Overby et al., [Pharmacological or genetic inhibition of Scn9a protects beta-cells while reducing insulin secretion in type 1 diabetes.](https://doi.org/10.1101/2023.06.11.544521) *bioRxiv* (2023). DOI: 10.1101/2023.06.11.544521 +- Mingze Dong et al., [Deep identifiable modeling of single-cell atlases enables zero-shot query of cellular states.](https://doi.org/10.1101/2023.11.11.566161) *bioRxiv* (2023). DOI: 10.1101/2023.11.11.566161 + +# 📰 News +- Documentary short film about *gget*: [https://youtu.be/cVR0k6Mt97o](https://youtu.be/cVR0k6Mt97o) +- Podcast episode for the Prototype Fund Public Interest Podcast about the importance of open-source software and its role in academic research (in German): [https://public-interest-podcast.podigee.io/33-pips4e4](https://public-interest-podcast.podigee.io/33-pips4e4) +- Prototype Fund announcement: [https://prototypefund.de/project/gget-genomische-datenbanken](https://prototypefund.de/project/gget-genomische-datenbanken/) diff --git a/docs/src/en/diamond.md b/docs/src/en/diamond.md index 7c829f6c..bccf89fa 100644 --- a/docs/src/en/diamond.md +++ b/docs/src/en/diamond.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget diamond 💎 +# gget diamond 💎 Align multiple protein or translated DNA sequences using [DIAMOND](https://www.nature.com/articles/nmeth.3176) (DIAMOND is similar to BLAST, but this is a local computation). Return format: JSON (command-line) or data frame/CSV (Python). @@ -10,6 +10,7 @@ Sequences (str or list) or path to FASTA file containing sequences to be aligned **Required arguments** `-ref` `--reference` Reference sequences (str or list) or path to FASTA file containing reference sequences. +Add the `--translated` flag (Python: `translated=True`) if reference sequences are amino acid sequences and query sequences are nucleotide sequences. **Optional arguments** `-db` `--diamond_db` @@ -30,6 +31,9 @@ Path to DIAMOND binary (str). Default: None -> Uses DIAMOND binary installed wit Path to the folder to save results in (str), e.g. "path/to/directory". Default: Standard out; temporary files are deleted. **Flags** +`-x` `--translated` +Perform translated alignment of nucleotide sequences to amino acid reference sequences. + `-csv` `--csv` Command-line only. Returns results in CSV format. Python: Use `json=True` to return output in JSON format. @@ -56,3 +60,10 @@ gget.diamond(["GGETISAWESQME", "ELVISISALIVE", "LQVEFRANKLIN", "PACHTERLABRQCKS" |Seq3 |Seq1 |100 |15 |15 |15 |0 |0 |1 |15 |1 |15 |2.01e-11|36.2 | #### [More examples](https://github.com/pachterlab/gget_examples) + +# References +If you use `gget diamond` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Buchfink, B., Xie, C. & Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015). [https://doi.org/10.1038/nmeth.3176](https://doi.org/10.1038/nmeth.3176) diff --git a/docs/src/en/elm.md b/docs/src/en/elm.md index d27228a7..4e810f2f 100644 --- a/docs/src/en/elm.md +++ b/docs/src/en/elm.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget elm 🎭 +# gget elm 🎭 Locally predict Eukaryotic Linear Motifs from an amino acid sequence or UniProt Acc using data from the [ELM database](http://elm.eu.org/). Return format: JSON (command-line) or data frame/CSV (Python). This module returns two data frames (or JSON formatted files) (see examples). @@ -7,10 +7,6 @@ Return format: JSON (command-line) or data frame/CSV (Python). This module retur Before using `gget elm` for the first time, run `gget setup elm` (bash) / `gget.setup("elm")` (Python) once (also see [`gget setup`](setup.md)). -If you use `gget elm` in a publication, please cite: -- Laura Luebbert, Chi Hoang, Manjeet Kumar, Lior Pachter, Fast and scalable querying of eukaryotic linear motifs with gget elm, _Bioinformatics_, 2024, btae095, [https://doi.org/10.1093/bioinformatics/btae095](https://doi.org/10.1093/bioinformatics/btae095) -- Manjeet Kumar, _et al._, The Eukaryotic Linear Motif resource: 2022 release, _Nucleic Acids Research_, Volume 50, Issue D1, 7 January 2022, Pages D497–D508, [https://doi.org/10.1093/nar/gkab975](https://doi.org/10.1093/nar/gkab975) - **Positional argument** `sequence` Amino acid sequence or Uniprot Acc (str). @@ -87,4 +83,16 @@ regex_df: |ELME000231 |DEG_APCC_DBOX_1 |APCC-binding Destruction motifs|DEG |An RxxL-based motif that binds to the Cdh1 and Cdc20 components of APC/C thereby targeting the protein for destruction in a cell cycle dependent manner|SRVKLNIVR |Saccharomyces cerevisiae S288c|… | |… |… |… |… |… |… |… |… | -#### [More examples](https://github.com/pachterlab/gget_examples) +# Tutorials +### [🔗 General `gget elm` demo](https://github.com/pachterlab/gget_examples/blob/main/gget_elm_demo.ipynb) + +### [🔗 A point mutation in BRCA2 is carcinogenic due to the loss of a protein interaction motif](https://github.com/pachterlab/gget_examples/blob/main/gget_elm_BRCA2_example.ipynb) + +### [🔗 Filter `gget elm` results based on disordered protein regions](https://github.com/pachterlab/gget_examples/blob/main/gget_elm_IUPred3_tutorial.ipynb) + +# References +If you use `gget elm` in a publication, please cite the following articles: + +- Laura Luebbert, Chi Hoang, Manjeet Kumar, Lior Pachter, Fast and scalable querying of eukaryotic linear motifs with gget elm, _Bioinformatics_, 2024, btae095, [https://doi.org/10.1093/bioinformatics/btae095](https://doi.org/10.1093/bioinformatics/btae095) + +- Manjeet Kumar, Sushama Michael, Jesús Alvarado-Valverde, Bálint Mészáros, Hugo Sámano‐Sánchez, András Zeke, Laszlo Dobson, Tamas Lazar, Mihkel Örd, Anurag Nagpal, Nazanin Farahi, Melanie Käser, Ramya Kraleti, Norman E Davey, Rita Pancsa, Lucía B Chemes, Toby J Gibson, The Eukaryotic Linear Motif resource: 2022 release, Nucleic Acids Research, Volume 50, Issue D1, 7 January 2022, Pages D497–D508, [https://doi.org/10.1093/nar/gkab975](https://doi.org/10.1093/nar/gkab975) diff --git a/docs/src/en/enrichr.md b/docs/src/en/enrichr.md index 3cf6bd3d..70f6abac 100644 --- a/docs/src/en/enrichr.md +++ b/docs/src/en/enrichr.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget enrichr 💰 +# gget enrichr 💰 Perform an enrichment analysis on a list of genes using [Enrichr](https://maayanlab.cloud/Enrichr/) or [modEnrichr](https://maayanlab.cloud/modEnrichr/). Return format: JSON (command-line) or data frame/CSV (Python). @@ -23,8 +23,8 @@ NOTE: database shortcuts are not supported for species other than 'human' or 'mo **Optional arguments** `-s` `--species` -Species to use as reference for the enrichment analysis. (Default: human) -Options: +Species to use as reference for the enrichment analysis. (Default: human) +Options: | Species | Database list | |----------|-------------------------------------------------------------------| @@ -220,4 +220,19 @@ df |> xlab("-log10(adjusted P value)") ``` -#### [More examples](https://github.com/pachterlab/gget_examples) +# Tutorials +[Using `gget enrichr` with background genes](https://github.com/pachterlab/gget_examples/blob/main/gget_enrichr_with_background_genes.ipynb) + +# References +If you use `gget enrichr` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 128(14). [https://doi.org/10.1186/1471-2105-14-128 ](https://doi.org/10.1186/1471-2105-14-128) + +- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 2016; gkw377. doi: [10.1093/nar/gkw377](https://doi.org/10.1093/nar/gkw377) + +- Xie Z, Bailey A, Kuleshov MV, Clarke DJB., Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, Jeon M, & Ma’ayan A. Gene set knowledge discovery with Enrichr. Current Protocols, 1, e90. 2021. doi: [10.1002/cpz1.90](https://doi.org/10.1002/cpz1.90). + +If working with non-human/mouse datasets, please also cite: +- Kuleshov MV, Diaz JEL, Flamholz ZN, Keenan AB, Lachmann A, Wojciechowicz ML, Cagan RL, Ma'ayan A. modEnrichr: a suite of gene set enrichment analysis tools for model organisms. Nucleic Acids Res. 2019 Jul 2;47(W1):W183-W190. doi: [10.1093/nar/gkz347](https://doi.org/10.1093/nar/gkz347). PMID: 31069376; PMCID: PMC6602483. diff --git a/docs/src/en/gpt.md b/docs/src/en/gpt.md index fa21bf9a..f90f0d3b 100644 --- a/docs/src/en/gpt.md +++ b/docs/src/en/gpt.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget gpt 💬 +# gget gpt 💬 Generates natural language text based on a given prompt using the [OpenAI](https://openai.com/) API's 'openai.ChatCompletion.create' endpoint. This module, including its source code, documentation and unit tests, were partly written by OpenAI's Chat-GTP3. diff --git a/docs/src/en/info.md b/docs/src/en/info.md index 5b73fffe..68684ec3 100644 --- a/docs/src/en/info.md +++ b/docs/src/en/info.md @@ -1,11 +1,12 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget info 💡 +# gget info 💡 Fetch extensive gene and transcript metadata from [Ensembl](https://www.ensembl.org/), [UniProt](https://www.uniprot.org/), and [NCBI](https://www.ncbi.nlm.nih.gov/) using Ensembl IDs. Return format: JSON (command-line) or data frame/CSV (Python). **Positional argument** `ens_ids` -One or more Ensembl IDs (WormBase and Flybase IDs are also supported). +One or more Ensembl IDs (WormBase and Flybase IDs are also supported). +NOTE: Providing a list of more than 1,000 Ensembl IDs at once might result in a server error (to process more than 1,000 IDs, split the list of IDs into chunks of 1,000 IDs and run these separately). **Optional arguments** `-o` `--out` @@ -53,3 +54,15 @@ gget.info(["ENSG00000034713", "ENSG00000104853", "ENSG00000170296"]) | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | ... | #### [More examples](https://github.com/pachterlab/gget_examples) + +# References +If you use `gget info` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. + +- Sayers EW, Beck J, Bolton EE, Brister JR, Chan J, Comeau DC, Connor R, DiCuccio M, Farrell CM, Feldgarden M, Fine AM, Funk K, Hatcher E, Hoeppner M, Kane M, Kannan S, Katz KS, Kelly C, Klimke W, Kim S, Kimchi A, Landrum M, Lathrop S, Lu Z, Malheiro A, Marchler-Bauer A, Murphy TD, Phan L, Prasad AB, Pujar S, Sawyer A, Schmieder E, Schneider VA, Schoch CL, Sharma S, Thibaud-Nissen F, Trawick BW, Venkatapathi T, Wang J, Pruitt KD, Sherry ST. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2024 Jan 5;52(D1):D33-D43. doi: [10.1093/nar/gkad1044](https://doi.org/10.1093/nar/gkad1044). PMID: 37994677; PMCID: PMC10767890. + +- The UniProt Consortium , UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Research, Volume 51, Issue D1, 6 January 2023, Pages D523–D531, [https://doi.org/10.1093/nar/gkac1052](https://doi.org/10.1093/nar/gkac1052) + diff --git a/docs/src/en/installation.md b/docs/src/en/installation.md index 46b5a3c0..7004741e 100644 --- a/docs/src/en/installation.md +++ b/docs/src/en/installation.md @@ -1,6 +1,6 @@ [](https://pypi.org/project/gget) [](https://anaconda.org/bioconda/gget) -## Installation +# Installation ```bash pip install --upgrade gget ``` diff --git a/docs/src/en/introduction.md b/docs/src/en/introduction.md index d8c8ce8d..7a02ea6d 100644 --- a/docs/src/en/introduction.md +++ b/docs/src/en/introduction.md @@ -15,35 +15,48 @@`gget` consists of a collection of separate but interoperable modules, each designed to facilitate one type of database querying in a single line of code.
-The databases queried by `gget` are continuously being updated which sometimes changes their structure. `gget` modules are tested automatically on a biweekly basis and updated to match new database structures when necessary. If you encounter a problem, please upgrade to the latest `gget` version using `pip install --upgrade gget`. If the problem persists, please [report the issue](https://github.com/pachterlab/gget/issues/new/choose). +NOTE: The databases queried by `gget` are continuously being updated which sometimes changes their structure. `gget` modules are tested automatically on a biweekly basis and updated to match new database structures when necessary. If you encounter a problem, please upgrade to the latest `gget` version using `pip install --upgrade gget`. If the problem persists, please [report the issue](https://github.com/pachterlab/gget/issues/new/choose).
[
Request a new feature
](https://github.com/pachterlab/gget/issues/new/choose)
> `gget info` and `gget seq` are currently unable to fetch information for WormBase and FlyBase IDs (all other IDs are functioning normally). This issue arose due to a bug in Ensembl release 112. We appreciate Ensembl's efforts in addressing this issue and expect a fix soon. Thank you for your patience. -
- -[
 ](/gget/en/alphafold.md)
-[
](/gget/en/alphafold.md)
-[ ](/gget/en/archs4.md)
-[
](/gget/en/archs4.md)
-[ ](/gget/en/blast.md)
-
-[
](/gget/en/blast.md)
-
-[ ](/gget/en/blat.md)
-[
](/gget/en/blat.md)
-[ ](/gget/en/cellxgene.md)
-[
](/gget/en/cellxgene.md)
-[ ](/gget/en/cosmic.md)
-[
](/gget/en/cosmic.md)
-[ ](/gget/en/diamond.md)
-[
](/gget/en/diamond.md)
-[ ](/gget/en/elm.md)
-[
](/gget/en/elm.md)
-[ ](/gget/en/enrichr.md)
+# gget modules
-[
](/gget/en/enrichr.md)
+# gget modules
-[ ](/gget/en/info.md)
-[
](/gget/en/info.md)
-[ ](/gget/en/muscle.md)
-[
](/gget/en/muscle.md)
-[ ](/gget/en/pdb.md)
+These are the `gget` core modules. Click on any module to access detailed documentation.
-[
](/gget/en/pdb.md)
+These are the `gget` core modules. Click on any module to access detailed documentation.
-[ ](/gget/en/ref.md)
-[
](/gget/en/ref.md)
-[ ](/gget/en/search.md)
-[
](/gget/en/search.md)
-[ ](/gget/en/seq.md)
-
-### [More tutorials](https://github.com/pachterlab/gget_examples)
+
](/gget/en/seq.md)
-
-### [More tutorials](https://github.com/pachterlab/gget_examples)
+| gget alphafold Predict 3D protein structure from an amino acid sequence. |
+ gget archs4 What is the expression of my gene in tissue X? |
+ gget bgee Find all orthologs of a gene. |
+ gget blast BLAST a nucleotide or amino acid sequence. |
+
| gget blat Find the genomic location of a nucleotide or amino acid sequence. |
+ gget cbio Explore a gene's expression in the specified cancers. |
+ gget cellxgene Get ready-to-use single-cell RNA seq count matrices from certain tissues/ diseases/ etc. |
+ gget cosmic Search for genes, mutations, and other factors associated with certain cancers. |
+
| gget diamond Align amino acid sequences to a reference. |
+ gget elm Find protein interaction domains and functions in an amino acid sequence. |
+ gget enrichr Check if a list of genes is associated with a specific celltype/ pathway/ disease/ etc. |
+ gget info Fetch all of the information associated with an Ensembl ID. |
+
| gget muscle Align multiple nucleotide or amino acid sequences to each other. |
+ gget mutate Mutate nucleotide sequences based on specified mutations. |
+ gget opentargets Explore which diseases and drugs a gene is associated with. |
+ gget pdb Fetch data from the Protein Data Bank (PDB) based on a PDB ID. |
+
| gget ref Get reference genomes from Ensembl. |
+ gget search Find Ensembl IDs associated with the specified search word. |
+ gget seq Fetch the nucleotide or amino acid sequence of a gene. |
+
@@ -56,5 +69,5 @@ Read the article here: [https://doi.org/10.1093/bioinformatics/btac836](https://
- - + + diff --git a/docs/src/en/muscle.md b/docs/src/en/muscle.md index f7be1aa6..cacf7168 100644 --- a/docs/src/en/muscle.md +++ b/docs/src/en/muscle.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget muscle 🦾 +# gget muscle 🦾 Align multiple nucleotide or amino acid sequences to each other using [Muscle5](https://www.drive5.com/muscle/). Return format: ClustalW formatted standard out or aligned FASTA (.afa). @@ -56,3 +56,11 @@ alv.view(msa) ``` #### [More examples](https://github.com/pachterlab/gget_examples) + +# References +If you use `gget muscle` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Edgar RC (2021), MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping, bioRxiv 2021.06.20.449169. [https://doi.org/10.1101/2021.06.20.449169](https://doi.org/10.1101/2021.06.20.449169) + diff --git a/docs/src/en/mutate.md b/docs/src/en/mutate.md index f175da88..316a1b78 100644 --- a/docs/src/en/mutate.md +++ b/docs/src/en/mutate.md @@ -5,6 +5,8 @@ Return format: Saves mutated sequences in FASTA format (or returns a list contai This module was written by [Joseph Rich](https://github.com/josephrich98). +** Update: The more complex functionality of gget mutate has been ported to https://github.com/pachterlab/kvar. kvar expands on this functionality in the context of screening for variants/mutations in sequencing data. If this sounds interesting to you, please check it out! ** + **Positional argument** `sequences` Path to the FASTA file containing the sequences to be mutated, e.g., 'path/to/seqs.fa'. @@ -53,60 +55,12 @@ Name of the column containing the IDs of the sequences to be mutated in `mutatio `-mic` `--mut_id_column` Name of the column containing the IDs of each mutation in `mutations`. Default: Same as `mut_column`. - -`-gtf` `--gtf` -Path to a .gtf file. When providing a genome fasta file as input for 'sequences', you can provide a .gtf file here and the input sequences will be defined according to the transcript boundaries, e.g. 'path/to/genome_annotation.gtf'. Default: None - -`-gtic` `--gtf_transcript_id_column` -Column name in the input `mutations` file containing the transcript ID. In this case, column `seq_id_column` should contain the chromosome number. -Required when `gtf` is provided. Default: None **Optional mutant sequence generation/filtering arguments** `-k` `--k` Length of sequences flanking the mutation. Default: 30. If k > total length of the sequence, the entire sequence will be kept. -`-msl` `--min_seq_len` -Minimum length of the mutant output sequence, e.g. 100. Mutant sequences smaller than this will be dropped. Default: None - -`-ma` `--max_ambiguous` -Maximum number of 'N' (or 'n') characters allowed in the output sequence, e.g. 10. Default: None (no ambiguous character filter will be applied) - -**Optional mutant sequence generation/filtering flags** -`-ofr` `--optimize_flanking_regions` -Removes nucleotides from either end of the mutant sequence to ensure (when possible) that the mutant sequence does not contain any k-mers also found in the wildtype/input sequence. - -`-rswk` `--remove_seqs_with_wt_kmers` -Removes output sequences where at least one k-mer is also present in the wildtype/input sequence in the same region. -When used with `--optimize_flanking_regions`, only sequences for which a wildtpye kmer is still present after optimization will be removed. - -`-mio` `--merge_identical_off` -Do not merge identical mutant sequences in the output (by default, identical sequences will be merged by concatenating the sequence headers for all identical sequences). - -**Optional arguments to generate additional output** -This output is activated using the `--update_df` flag and will be stored in a copy of the `mutations` DataFrame. - -`-udf_o` `--update_df_out` -Path to output csv file containing the updated DataFrame, e.g. 'path/to/mutations_updated.csv'. Only valid when used with `--update_df`. -Default: None -> the new csv file will be saved in the same directory as the `mutations` DataFrame with appendix '_updated' - -`-ts` `--translate_start` -(int or str) The position in the input nucleotide sequence to start translating, e.g. 5. If a string is provided, it should correspond to a column name in `mutations` containing the open reading frame start positions for each sequence/mutation. Only valid when used with `--translate`. -Default: translates from the beginning of each sequence - -`-te` `--translate_end` -(int or str) The position in the input nucleotide sequence to end translating, e.g. 35. If a string is provided, it should correspond to a column name in `mutations` containing the open reading frame end positions for each sequence/mutation. Only valid when used with `--translate`. -Default: translates until the end of each sequence - -**Optional flags to modify additional output** -`-udf` `--update_df` -Updates the input `mutations` DataFrame to include additional columns with the mutation type, wildtype nucleotide sequence, and mutant nucleotide sequence (only valid if `mutations` is a .csv or .tsv file). - -`-sfs` `--store_full_sequences` -Includes the complete wildtype and mutant sequences in the updated `mutations` DataFrame (not just the sub-sequence with k-length flanks). Only valid when used with `--update_df`. - -`-tr` `--translate` -Adds additional columns to the updated `mutations` DataFrame containing the wildtype and mutant amino acid sequences. Only valid when used with `--store_full_sequences`. **Optional general arguments** `-o` `--out` @@ -160,39 +114,4 @@ gget.mutate(["ATCGCTAAGCT", "TAGCTA"], "c.1_3inv", k=3) → Returns ['CTAGCT', 'GATCTA']. -
- -**Pass in the genome mutation information as a `mutations` CSV (by having `seq_id_column` contain chromosome information, and `mut_column` contain mutation information with respect to genome coordinates), as well as the genome as the `sequences` file. Respect the transcript boundaries by merging in transcript start and end positions with the `gtf` argument set to the path to the gtf file, as well as the `gtf_transcript_id_column` specifying the name of the column containing transcript ID's corresponding to the gtf in the input `mutations` file. Optimize the length to maximize length while maintaining specificity of all k-mers with the `optimize_flanking_regions` argument. Create a CSV file with updated information including mutation type and output sequences with the `update_df argument`, stored to the path designated by `update_df_out`. Store the full sequences (i.e., the mutation in the context of the entire sequence of the corresponding `sequences` fasta file entry) with the `store_full_sequences` argument. Store translated amino acid sequences for each full mutation with the `translate` argument, with `translate_start` and `translate_end` specifying the names of the column in the input `mutations` file that contain the start and end sequence positions of the open reading frame, respectively:** -```bash -gget mutate genome_reference.fa -m mutations_input.csv -o mut_fasta.fa -k 4 -sic Chromosome -mic Mutation -gtf genome_annotation.gtf -gtic Ensembl_Transcript_ID -ofr -update_df -udf_o mutations_updated.csv -sfs -tr -ts Translate_Start -te Translate_End -``` -```python -# Python -gget.mutate(sequences="genome_reference.fa", mutations="mutations_input.csv", out="mut_fasta.fa", k=4, seq_id_column="Chromosome", mut_column="Mutation", gtf="genome_annotation.gtf", gtf_transcript_id_column="Ensembl_Transcript_ID", optimize_flanking_regions=True, update_df=True, update_df_out="mutations_updated.csv", store_full_sequences=True, translate=True, translate_start="Translate_Start", translate_end="Translate_End") -``` -→ Takes as input 'mutations_input.csv' file containing: -``` -| Chromosome | Mutation | Ensembl_Transcript_ID | Translate_Start | Translate_End | -|------------|-------------------|------------------------|-----------------|---------------| -| 1 | g.224411A>C | ENST00000193812 | 0 | 100 | -| 8 | g.25111del | ENST00000174411 | 0 | 294 | -| X | g.1011_1012insAA | ENST00000421914 | 9 | 1211 | -``` -→ Saves 'mut_fasta.fa' file containing: -``` ->1:g.224411A>C -TGCTCTGCT ->8:g.25111del -GAGTCGAT ->X:g.1011_1012insAA -TTAGAACTT -``` -→ Saves 'mutations_updated.csv' file containing: -``` - -| Chromosome | Mutation | Ensembl_Transcript_ID | mutation_type | wt_sequence | mutant_sequence | wt_sequence_full | mutant_sequence_full | wt_sequence_aa_full | mutant_sequence_aa_full | -|------------|-------------------|------------------------|---------------|-------------|-----------------|-------------------|----------------------|---------------------|-------------------------| -| 1 | g.224411A>C | ENSMUST00000193812 | Substitution | TGCTATGCT | TGCTCTGCT | ...TGCTATGCT... | ...TGCTCTGCT... | ...CYA... | ...CSA... | -| 8 | g.25111del | ENST00000174411 | Deletion | GAGTCCGAT | GAGTCGAT | ...GAGTCCGAT... | ...GAGTCGAT... | ...ESD... | ...ES... | -| X | g.1011_1012insAA | ENST00000421914 | Insertion | TTAGCTT | TTAGAACTT | ...TTAGCTT... | ...TTAGAACTT... | ...A... | ...EL... | - +
\ No newline at end of file diff --git a/docs/src/en/opentargets.md b/docs/src/en/opentargets.md index f2d7f7c8..6e431076 100644 --- a/docs/src/en/opentargets.md +++ b/docs/src/en/opentargets.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget opentargets 🎯 +# gget opentargets 🎯 Fetch associated diseases or drugs from [OpenTargets](https://platform.opentargets.org/) using Ensembl IDs. Return format: JSON/CSV (command-line) or data frame (Python). @@ -270,3 +270,11 @@ gget.opentargets( #### [More examples](https://github.com/pachterlab/gget_examples) + +# References +If you use `gget opentargets` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Ochoa D, Hercules A, Carmona M, Suveges D, Baker J, Malangone C, Lopez I, Miranda A, Cruz-Castillo C, Fumis L, Bernal-Llinares M, Tsukanov K, Cornu H, Tsirigos K, Razuvayevskaya O, Buniello A, Schwartzentruber J, Karim M, Ariano B, Martinez Osorio RE, Ferrer J, Ge X, Machlitt-Northen S, Gonzalez-Uriarte A, Saha S, Tirunagari S, Mehta C, Roldán-Romero JM, Horswell S, Young S, Ghoussaini M, Hulcoop DG, Dunham I, McDonagh EM. The next-generation Open Targets Platform: reimagined, redesigned, rebuilt. Nucleic Acids Res. 2023 Jan 6;51(D1):D1353-D1359. doi: [10.1093/nar/gkac1046](https://doi.org/10.1093/nar/gkac1046). PMID: 36399499; PMCID: PMC9825572. + diff --git a/docs/src/en/pdb.md b/docs/src/en/pdb.md index c9b64db4..044d6407 100644 --- a/docs/src/en/pdb.md +++ b/docs/src/en/pdb.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget pdb 🔮 +# gget pdb 🔮 Query [RCSB Protein Data Bank (PDB)](https://www.rcsb.org/) for the protein structure/metadata of a given PDB ID. Return format: Resource 'pdb' is returned in PDB format. All other resources are returned in JSON format. @@ -78,3 +78,12 @@ gget.pdb("7CT5", save=True) → The use case above exemplifies how to find PDB files for comparative analysis of protein structure starting with Ensembl IDs or amino acid sequences. The fetched PDB files can also be compared to predicted structures generated by [`gget alphafold`](alphafold.md). PDB files can be viewed interactively in 3D [online](https://rcsb.org/3d-view), or using programs like [PyMOL](https://pymol.org/) or [Blender](https://www.blender.org/). To compare two PDB files, you can use [this website](https://rcsb.org/alignment). #### [More examples](https://github.com/pachterlab/gget_examples) + +# References +If you use `gget pdb` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000 Jan 1;28(1):235-42. doi: [10.1093/nar/28.1.235](https://doi.org/10.1093/nar/28.1.235). PMID: 10592235; PMCID: PMC102472. + + diff --git a/docs/src/en/quick_start_guide.md b/docs/src/en/quick_start_guide.md index 5c29622f..7bd931ba 100644 --- a/docs/src/en/quick_start_guide.md +++ b/docs/src/en/quick_start_guide.md @@ -1,4 +1,4 @@ -## 🪄 Quick start guide +# 🪄 Quick start guide Command line: ```bash # Fetch all Homo sapiens reference and annotation FTPs from the latest Ensembl release @@ -90,3 +90,4 @@ gget$archs4("ACE2", which="tissue") gget$pdb("1R42", save=TRUE) ``` #### [More examples](https://github.com/pachterlab/gget_examples) + diff --git a/docs/src/en/ref.md b/docs/src/en/ref.md index e81bbccf..889109dd 100644 --- a/docs/src/en/ref.md +++ b/docs/src/en/ref.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget ref 📖 +# gget ref 📖 Fetch FTPs and their respective metadata (or use flag `ftp` to only return the links) for reference genomes and annotations from [Ensembl](https://www.ensembl.org/) by species. Return format: dictionary/JSON. @@ -50,26 +50,6 @@ Python: Use `verbose=False` to prevent progress information from being displayed ### Examples -**Use `gget ref` in combination with [kallisto | bustools](https://www.kallistobus.tools/kb_usage/kb_ref/) to build a reference index:** -```bash -kb ref -i INDEX -g T2G -f1 FASTA $(gget ref --ftp -w dna,gtf homo_sapiens) -``` -→ kb ref builds a reference index using the latest DNA and GTF files of species **Homo sapiens** passed to it by `gget ref`. - -
- -**List all available genomes from Ensembl release 103:** -```bash -gget ref --list_species -r 103 -``` -```python -# Python -gget.ref(species=None, list_species=True, release=103) -``` -→ Returns a list with all available genomes (checks if GTF and FASTAs are available) from Ensembl release 103. -(If no release is specified, `gget ref` will always return information from the latest Ensembl release.) - -
**Get the genome reference for a specific species:** ```bash @@ -101,4 +81,36 @@ gget.ref("homo_sapiens", which=["gtf", "dna"]) } ``` +
+ +**List all available genomes from Ensembl release 103:** +```bash +gget ref --list_species -r 103 +``` +```python +# Python +gget.ref(species=None, list_species=True, release=103) +``` +→ Returns a list with all available genomes (checks if GTF and FASTAs are available) from Ensembl release 103. +(If no release is specified, `gget ref` will always return information from the latest Ensembl release.) + +
+ +**Use `gget ref` in combination with [kallisto | bustools](https://www.kallistobus.tools/kb_usage/kb_ref/) to build a reference index:** +```bash +kb ref \ + -i index.idx \ + -g t2g.txt \ + -f1 fasta.fa \ + $(gget ref --ftp -w dna,gtf homo_sapiens) +``` +→ kb ref builds a reference index using the latest DNA and GTF files of species **Homo sapiens** passed to it by `gget ref`. + #### [More examples](https://github.com/pachterlab/gget_examples) + +# References +If you use `gget ref` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. diff --git a/docs/src/en/search.md b/docs/src/en/search.md index c58262f6..6321a7cd 100644 --- a/docs/src/en/search.md +++ b/docs/src/en/search.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget search 🔎 +# gget search 🔎 Fetch genes and transcripts from [Ensembl](https://www.ensembl.org/) using free-form search terms. Results are matched based on the "gene name" and "description" sections in the Ensembl database. `gget` version >= 0.27.9 also includes results that match the Ensembl "synonym" section. Return format: JSON (command-line) or data frame/CSV (Python). @@ -74,3 +74,11 @@ gget.search(["gaba", "gamma-aminobutyric"], "homo_sapiens") | . . . | . . . | . . . | . . . | . . . | . . . | #### [More examples](https://github.com/pachterlab/gget_examples) + +# References +If you use `gget search` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. + diff --git a/docs/src/en/seq.md b/docs/src/en/seq.md index fd13eb3a..b1b13830 100644 --- a/docs/src/en/seq.md +++ b/docs/src/en/seq.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget seq 🧬 +# gget seq 🧬 Fetch nucleotide or amino acid sequence(s) of a gene (and all its isoforms) or a transcript by Ensembl ID. Return format: FASTA. @@ -48,3 +48,12 @@ gget.seq("ENSG00000034713", translate=True, isoforms=True) → Returns the amino acid sequences of all known transcripts of ENSG00000034713 in FASTA format. #### [More examples](https://github.com/pachterlab/gget_examples) + +# References +If you use `gget seq` in a publication, please cite the following articles: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606 + +- The UniProt Consortium , UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Research, Volume 51, Issue D1, 6 January 2023, Pages D523–D531, [https://doi.org/10.1093/nar/gkac1052](https://doi.org/10.1093/nar/gkac1052) diff --git a/docs/src/en/setup.md b/docs/src/en/setup.md index 078c869b..0d1482aa 100644 --- a/docs/src/en/setup.md +++ b/docs/src/en/setup.md @@ -1,5 +1,5 @@ > Python arguments are equivalent to long-option arguments (`--arg`), unless otherwise specified. Flags are True/False arguments in Python. The manual for any gget tool can be called from the command-line using the `-h` `--help` flag. -## gget setup 🔧 +# gget setup 🔧 Function to install/download third-party dependencies for a specified gget module. diff --git a/docs/src/en/updates.md b/docs/src/en/updates.md index 2e5d17fa..5ce7aaea 100644 --- a/docs/src/en/updates.md +++ b/docs/src/en/updates.md @@ -1,32 +1,4 @@ ## ✨ What's new -**Version ≥ 0.29.0** (Sep 25, 2024): -- **New modules:** - - [`gget cbio`](cbio.md) - - [`gget opentargets`](opentargets.md) - - [`gget bgee`](bgee.md) -- **[`gget enrichr`](./enrichr.md) now also supports species other than human (fly, yeast, worm, and fish) via [modEnrichR](https://maayanlab.cloud/modEnrichr/)** -- [`gget mutate`](./mutate.md): - `gget mutate` will now merge identical sequences in the final file by default. Mutation creation was vectorized to decrease runtime. Improved flanking sequence check for non-substitution mutations to make sure no wildtype kmer is retained in the mutation-containing sequence. Addition of several new arguments to customize sequence generation and output. -- [`gget cosmic`](./cosmic.md): - Added support for targeted as well as gene screens. The CSV file created for gget mutate now also contains protein mutation info. -- [`gget ref`](./ref.md): - Added out file option. -- [`gget info`](./info.md) and [`gget seq`](./seq.md): - Switched to Ensembl POST API to increase speed (nothing changes in front end). -- Other "behind the scenes" changes: - - Unit tests reorganized to increase speed and decrease code - - Requirements updated to [allow newer mysql-connector versions](https://github.com/pachterlab/gget/pull/159) - - [Support Numpy>= 2.0](https://github.com/pachterlab/gget/issues/157) - -**Version ≥ 0.28.6** (Jun 2, 2024): -- **New module: [`gget mutate`](./mutate.md)** -- [`gget cosmic`](./cosmic.md): You can now download entire COSMIC databases using the argument `download_cosmic` argument -- [`gget ref`](./ref.md): Can now fetch the GRCh37 genome assembly using `species='human_grch37'` -- [`gget search`](./search.md): Adjust access of human data to the structure of Ensembl release 112 (fixes [issue 129](https://github.com/pachterlab/gget/issues/129)) - -~~**Version ≥ 0.28.5** (May 29, 2024):~~ -- Yanked due to logging bug in `gget.setup("alphafold")` + inversion mutations in `gget mutate` only reverse the string instead of also computing the complementary strand - **Version ≥ 0.28.4** (January 31, 2024): - [`gget setup`](./setup.md): Fix bug with filepath when running `gget.setup("elm")` on Windows OS. @@ -41,7 +13,7 @@ - The regex string for regular expression matches was encapsulated as follows: "(?=(regex))" (instead of directly passing the regex string "regex") to enable capturing all occurrences of a motif when the motif length is variable and there are repeats in the sequence ([https://regex101.com/r/HUWLlZ/1](https://regex101.com/r/HUWLlZ/1)). - [`gget setup`](./setup.md): Use the `out` argument to specify a directory the ELM database will be downloaded into. Completes [this feature request](https://github.com/pachterlab/gget/issues/119). - [`gget diamond`](./diamond.md): The DIAMOND command is now run with `--ignore-warnings` flag, allowing niche sequences such as amino acid sequences that only contain nucleotide characters and repeated sequences. This is also true for DIAMOND alignments performed within [`gget elm`](./elm.md). -- **[`gget ref`](./ref.md) and [`gget search`](./search.md) back-end change: the current Ensembl release is fetched from the new [release file](https://ftp.ensembl.org/pub/VERSION) on the Ensembl FTP site to avoid errors during uploads of new releases.** +- [`gget ref`](./ref.md) and [`gget search`](./search.md) back-end change: the current Ensembl release is fetched from the new [release file](https://ftp.ensembl.org/pub/VERSION) on the Ensembl FTP site to avoid errors during uploads of new releases. - [`gget search`](./search.md): - FTP link results (`--ftp`) are saved in txt file format instead of json. - Fix URL links to Ensembl gene summary for species with a subspecies name and invertebrates. diff --git a/docs/src/es/alphafold.md b/docs/src/es/alphafold.md index c7ff56a2..300b37a0 100644 --- a/docs/src/es/alphafold.md +++ b/docs/src/es/alphafold.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget alphafold 🪢 +# gget alphafold 🪢 Predice la estructura en 3D de cualquier proteína derivada de su secuencia de aminoácidos usando una versión simplificada del algoritmo [AlphaFold2](https://github.com/deepmind/alphafold) de [DeepMind](https://www.deepmind.com/), originalmente producido y publicado para [AlphaFold Colab](https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb). Resultado: Predicción de la estructura (en formato PDB) y el errór de alineación (en formato json). @@ -68,5 +68,19 @@ gget.pdb("2K42", save=True) -### [Ejemplo en Google Colab](https://github.com/pachterlab/gget_examplblob/main/gget_alphafold.ipynb) -### [gget alphafold - preguntas más frecuentes](https://github.com/pachterlab/gget/discussions/39) +# Tutoriales +### [🔗 Google Colab tutorial](https://github.com/pachterlab/gget_examples/blob/main/gget_alphafold.ipynb) + +### [🔗 Predicción de la estructura de proteínas con comparación con estructuras cristalinas relacionadas](https://github.com/pachterlab/gget_examples/blob/main/protein_structure_prediction_comparison.ipynb) + +### [🔗 gget alphafold - preguntas más frecuentes](https://github.com/pachterlab/gget/discussions/39) + +# Citar +Si utiliza `gget alphafold` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [https://doi.org/10.1038/s41586-021-03819-2](https://doi.org/10.1038/s41586-021-03819-2) + +Y, si corresponde: +- Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021.10.04.463034; [https://doi.org/10.1101/2021.10.04.463034](https://doi.org/10.1101/2021.10.04.463034) diff --git a/docs/src/es/archs4.md b/docs/src/es/archs4.md index d9ec43ea..7407c7eb 100644 --- a/docs/src/es/archs4.md +++ b/docs/src/es/archs4.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no es especificado de otra manera. Las banderas son designadas como cierto o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede obtener desde Terminal con la bandera `-h` `--help`. -## gget archs4 🐁 +# gget archs4 🐁 Encuentra los genes más correlacionados a un gen de interés, o bién, encuentra los tejidos donde un gen se expresa usando la base de datos [ARCHS4](https://maayanlab.cloud/archs4/). Produce: Resultados en formato JSON (Terminal) o Dataframe/CSV (Python). @@ -76,3 +76,12 @@ Consulte [este tutorial](https://davetang.org/muse/2023/05/16/check-where-a-gene #### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget archs4` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, Wang L, Silverstein MC, Ma’ayan A. Massive mining of publicly available RNA-seq data from human and mouse. Nature Communications 9. Article number: 1366 (2018), doi:10.1038/s41467-018-03751-6 + +- Bray NL, Pimentel H, Melsted P and Pachter L, Near optimal probabilistic RNA-seq quantification, Nature Biotechnology 34, p 525--527 (2016). [https://doi.org/10.1038/nbt.3519](https://doi.org/10.1038/nbt.3519) diff --git a/docs/src/es/bgee.md b/docs/src/es/bgee.md new file mode 100644 index 00000000..73793ae0 --- /dev/null +++ b/docs/src/es/bgee.md @@ -0,0 +1,89 @@ + +> Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. +# gget bgee 🐝 + +Obtenga datos de ortología y expresión genética de [Bgee](https://www.bgee.org/) utilizando IDs de Ensembl. +Resultado: JSON/CSV (línea de comandos) o marco de datos (Python). + +> Si estás interesado específicamente en datos de expresión génica humana, considera usar [gget opentargets](./opentargets.md) o [gget archs4](./archs4.md) en su lugar. **gget bgee** tiene menos datos, pero admite más especies. + +Este módulo fue escrito por [Sam Wagenaar](https://github.com/techno-sam). + +**Argumento posicional** +`ens_id` +ID de gen Ensembl, por ejemplo, ENSG00000169194 o ENSSSCG00000014725. + +NOTA: Algunas de las especies en [Bgee](https://www.bgee.org/) no están en Ensembl, y para ellas puede utilizar los ID de genes del NCBI, p. 118215821 (un gen en _Anguilla anguilla_). + +**Argumentos requeridos** +`-t` `--type` +Tipo de datos a obtener. Opciones: `orthologs`, `expression`. + +**Argumentos opcionales** +`-o` `--out` +Ruta al archivo JSON donde se guardarán los resultados, por ejemplo, path/to/directory/results.json. Por defecto: Salida estándar. + +**Banderas** +`-csv` `--csv` +Solo en línea de comandos. Devuelve la salida en formato CSV, en lugar de formato JSON. +Python: Usa `json=True` para devolver la salida en formato JSON. + +`-q` `--quiet` +Solo en línea de comandos. Evita que se muestre la información de progreso. +Python: Usa `verbose=False` para evitar que se muestre la información de progreso. + +### Ejemplos + +**Obtener ortólogos para un gen** + +```bash +gget bgee ENSSSCG00000014725 -t orthologs +``` +```python +import gget +gget.bgee("ENSSSCG00000014725", type="orthologs") +``` + +→ Devuelve ortólogos para el gen con el ID de Ensembl ENSSSCG00000014725. + +| gene_id | gene_name | species_id | genus | species | +|--------------------|--------------|------------|---------|------------| +| 734881 | hbb1 | 8355 | Xenopus | laevis | +| ENSFCAG00000038029 | LOC101098159 | 9685 | Felis | catus | +| ENSBTAG00000047356 | LOC107131172 | 9913 | Bos | taurus | +| ENSOARG00000019163 | LOC101105437 | 9940 | Ovis | aries | +| ENSXETG00000025667 | hbg1 | 8364 | Xenopus | tropicalis | +| ... | ... | ... | ... | ... | + +
+ +**Obtener datos de expresión génica para un gen** + +```bash +gget bgee ENSSSCG00000014725 -t expression +``` +```python +import gget +gget.bgee("ENSSSCG00000014725", type="expression") +``` + +→ Devuelve datos de expresión génica para el gen con el ID de Ensembl ENSSSCG00000014725. + +| anat_entity_id | anat_entity_name | score | score_confidence | expression_state | +|----------------|-----------------------------|-------|------------------|------------------| +| UBERON:0000178 | blood | 99.98 | high | expressed | +| UBERON:0002106 | spleen | 99.96 | high | expressed | +| UBERON:0002190 | subcutaneous adipose tissue | 99.70 | high | expressed | +| UBERON:0005316 | endocardial endothelium | 99.61 | high | expressed | +| UBERON:0002107 | liver | 99.27 | high | expressed | +| ... | ... | ... | ... | ... | + + +#### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget bgee` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Frederic B Bastian, Julien Roux, Anne Niknejad, Aurélie Comte, Sara S Fonseca Costa, Tarcisio Mendes de Farias, Sébastien Moretti, Gilles Parmentier, Valentine Rech de Laval, Marta Rosikiewicz, Julien Wollbrett, Amina Echchiki, Angélique Escoriza, Walid H Gharib, Mar Gonzales-Porta, Yohan Jarosz, Balazs Laurenczy, Philippe Moret, Emilie Person, Patrick Roelli, Komal Sanjeev, Mathieu Seppey, Marc Robinson-Rechavi (2021). The Bgee suite: integrated curated expression atlas and comparative transcriptomics in animals. Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D831–D847, [https://doi.org/10.1093/nar/gkaa793](https://doi.org/10.1093/nar/gkaa793) diff --git a/docs/src/es/blast.md b/docs/src/es/blast.md index 4fbf8302..93066d28 100644 --- a/docs/src/es/blast.md +++ b/docs/src/es/blast.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Las banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget blast 💥 +# gget blast 💥 BLAST una secuencia de nucleótidos o aminoácidos a cualquier base de datos [BLAST](https://blast.ncbi.nlm.nih.gov/Blast.cgi). Produce: Resultados en formato JSON (Terminal) o Dataframe/CSV (Python). @@ -72,3 +72,10 @@ gget.blast("fasta.fa") → Produce los resultados BLAST de la primera secuencia contenida en el archivo 'fasta.fa'. #### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget blast` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990 Oct 5;215(3):403-10. doi: 10.1016/S0022-2836(05)80360-2. PMID: 2231712. diff --git a/docs/src/es/blat.md b/docs/src/es/blat.md index e4a498df..13e9f07b 100644 --- a/docs/src/es/blat.md +++ b/docs/src/es/blat.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget blat 🎯 +# gget blat 🎯 Encuentra la ubicación genómica de una secuencia de nucleótidos o aminoácidos usando [BLAT](https://genome.ucsc.edu/cgi-bin/hgBlat). Produce: Resultados en formato JSON (Terminal) o Dataframe/CSV (Python). @@ -45,3 +45,10 @@ gget.blat("MKWMFKEDHSLEHRCVESAKIRAKYPDRVPVIVEKVSGSQIVDIDKRKYLVPSDITVAQFMWIIRKRIQ | taeGut2| 88 | 12 | 88 | 77 | 0 | 87.5 | ... | #### [Màs ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget blat` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002 Apr;12(4):656-64. doi: 10.1101/gr.229202. PMID: 11932250; PMCID: PMC187518. diff --git a/docs/src/es/cbio.md b/docs/src/es/cbio.md new file mode 100644 index 00000000..f4e0683a --- /dev/null +++ b/docs/src/es/cbio.md @@ -0,0 +1,233 @@ +> Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. +# gget cbio 📖 + +Trazar mapas de calor de la genómica del cáncer utilizando datos de [cBioPortal](https://www.cbioportal.org/) con IDs de Ensembl o nombres de genes. + +Este módulo fue escrito por [Sam Wagenaar](https://github.com/techno-sam). + +**Argumento posicional** +`subcommand` +O bien `search` o `plot` + +### Subcomando `search` (Python: `gget.cbio_search`) +Buscar IDs de estudios de cBioPortal por palabra clave. +Formato de retorno: JSON (línea de comandos) o lista de cadenas (Python). +**Nota: Esto no devuelve estudios con tipos de cáncer mixtos.** + +**Argumento posicional** +`keywords` +Lista de palabras clave separadas por espacios para buscar, por ejemplo
breast lung.
+Python: Pasa palabras clave como una lista de cadenas.
+
+### Subcomando `plot` (Python: `gget.cbio_plot`)
+Graficar mapas de calor de genómica del cáncer utilizando datos de cBioPortal.
+Formato de retorno: PNG (línea de comandos y Python).
+
+**Argumentos requeridos**
+`-s` `--study_ids`
+Lista separada por espacios de IDs de estudios de cBioPortal, por ejemplo, msk_impact_2017 egc_msk_2023.
+
+`-g` `--genes`
+Lista separada por espacios de nombres de genes o IDs de Ensembl, por ejemplo, NOTCH3 ENSG00000108375.
+
+**Argumentos opcionales**
+`-st` `--stratification`
+Columna por la cual estratificar los datos. Predeterminado: `tissue`.
+Opciones:
+- tissue
+- cancer_type
+- cancer_type_detailed
+- study_id
+- sample
+
+`-vt` `--variation_type`
+Tipo de variación a graficar. Predeterminado: `mutation_occurrences`.
+Opciones:
+- mutation_occurrences
+- cna_nonbinary (Nota: la `stratification` debe ser 'sample' para esta opción)
+- sv_occurrences
+- cna_occurrences
+- Consequence (Nota: la `stratification` debe ser 'sample' para esta opción)
+
+`-f` `--filter`
+Filtrar los datos por un valor específico en una columna específica, por ejemplo, `study_id:msk_impact_2017`.
+Python: `filter=(column, value)`
+
+`-dd` `--data_dir`
+Directorio para almacenar los archivos de datos. Predeterminado: `./gget_cbio_cache`.
+
+`-fd` `--figure_dir`
+Directorio para las figuras de salida. Predeterminado: `./gget_cbio_figures`.
+
+`-fn` `--filename`
+Nombre del archivo de salida, relativo a `figure_dir`. Predeterminado: auto-generado.
+Python: `figure_filename`.
+
+`-t` `--title`
+Título para la figura de salida. Predeterminado: auto-generado.
+Python: `figure_title`.
+
+`-dpi` `--dpi`
+DPI de la figura de salida. Predeterminado: 100.
+
+**Banderas**
+
+`-q` `--quiet`
+Solo en línea de comandos. Evita que se muestre la información de progreso.
+Python: Usa `verbose=False` para evitar que se muestre la información de progreso.
+
+`-nc` `--no_confirm`
+Solo en línea de comandos. Omitir las confirmaciones de descarga.
+Python: Usa `confirm_download=True` para habilitar las confirmaciones de descarga.
+
+`-sh` `--show`
+Mostrar la gráfica en una ventana (automático en notebooks de Jupyter).
+
+### Ejemplos
+
+**Encontrar todos los estudios de cBioPortal con tipos de cáncer que coinciden con palabras clave específicas:**
+```bash
+gget cbio search esophag ovary ovarian
+```
+```python
+# Python
+import gget
+gget.cbio_search(['esophag', 'ovary', 'ovarian'])
+```
+→ Devuelve una lista de estudios con tipos de cáncer que coinciden con las palabras clave `esophag`, `ovary`, o `ovarian`.
+
+```
+['egc_tmucih_2015', 'egc_msk_2017', ..., 'msk_spectrum_tme_2022']
+```
+
++ +**Graficar un mapa de calor de ocurrencias de mutaciones para genes específicos en un estudio específico:** +```bash +gget cbio plot \ + -s msk_impact_2017 \ + -g AKT1 ALK FLT4 MAP3K1 MLL2 MLL3 NOTCH3 NOTCH4 PDCD1 RNF43 \ + -st tissue \ + -vt mutation_occurrences \ + -dpi 200 +``` +```python +# Python +import gget +gget.cbio_plot( + ['msk_impact_2017'], + ['AKT1', 'ALK', 'FLT4', 'MAP3K1', 'MLL2', 'MLL3', 'NOTCH3', 'NOTCH4', 'PDCD1', 'RNF43'], + stratification='tissue', + variation_type='mutation_occurrences', + dpi=200 +) +``` + +→ Guarda un mapa de calor de ocurrencias de mutaciones para los genes especificados en el estudio especificado en ./gget_cbio_figures/Heatmap_tissue.png. + +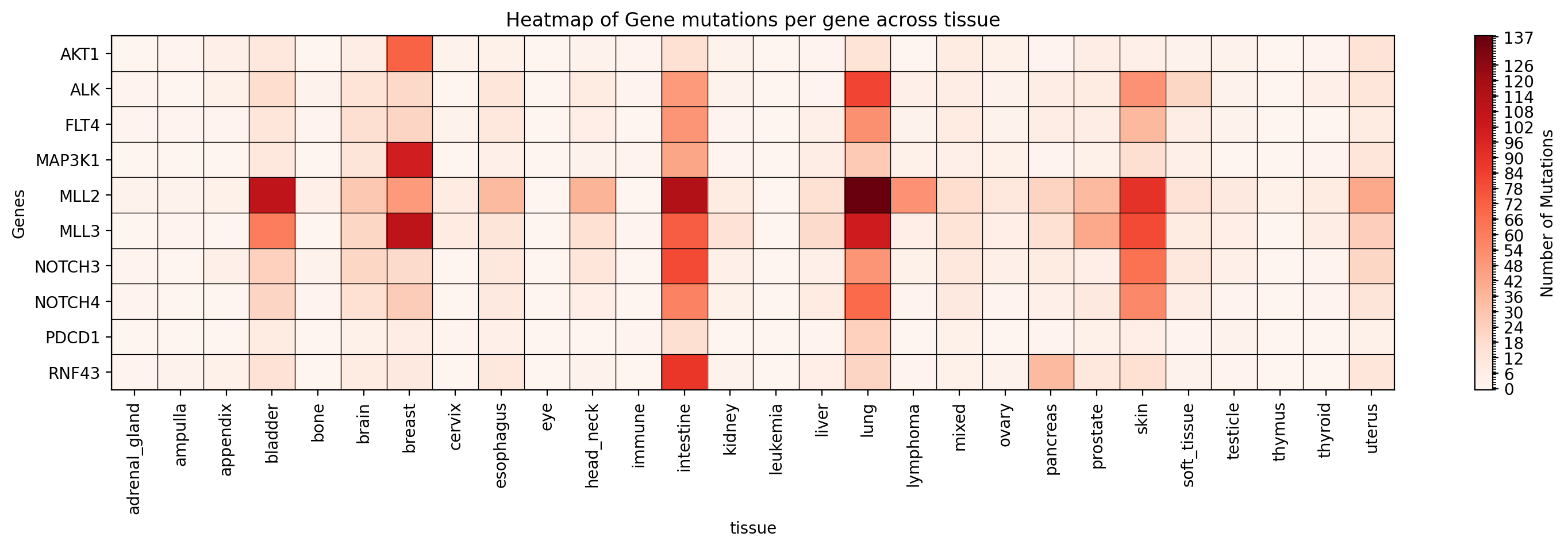 + +
+ +**Graficar un mapa de calor de tipos de mutaciones para genes específicos en un estudio específico:** +```bash +gget cbio plot \ + -s msk_impact_2017 \ + -g AKT1 ALK FLT4 MAP3K1 MLL2 MLL3 NOTCH3 NOTCH4 PDCD1 RNF43 \ + -st sample \ + -vt Consequence \ + -dpi 200 +``` +```python +# Python +import gget +gget.cbio_plot( + ['msk_impact_2017'], + ['AKT1', 'ALK', 'FLT4', 'MAP3K1', 'MLL2', 'MLL3', 'NOTCH3', 'NOTCH4', 'PDCD1', 'RNF43'], + stratification='sample', + variation_type='Consequence', + dpi=200, +) +``` + +→ Guarda un mapa de calor de tipos de mutaciones para los genes especificados en el estudio especificado en ./gget_cbio_figures/Heatmap_sample.png. + +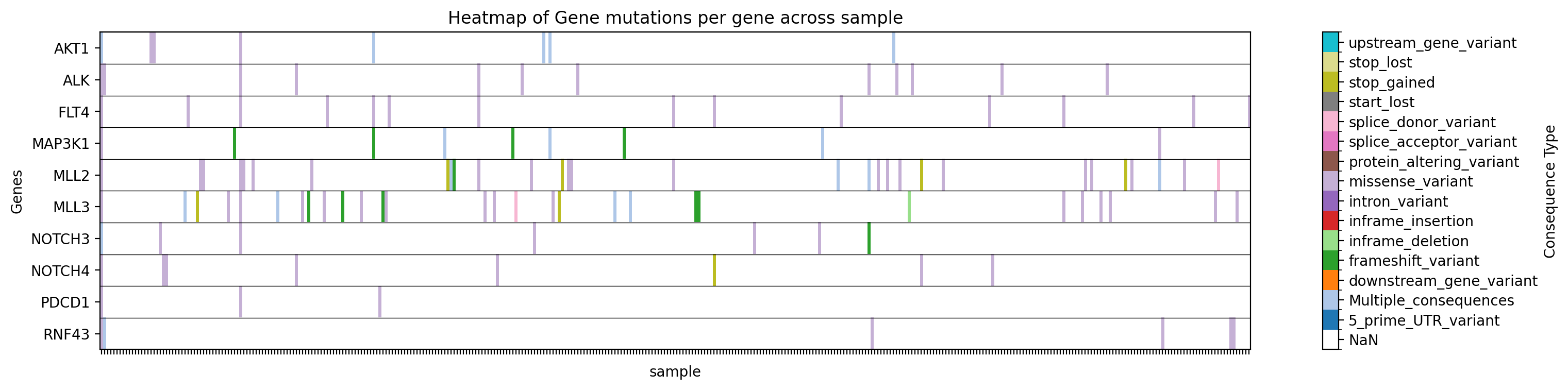 + +
+ +**Graficar un mapa de calor de tipos de mutaciones para genes específicos en un estudio específico, filtrando por tejido::** +```bash +gget cbio plot \ + -s msk_impact_2017 \ + -g AKT1 ALK FLT4 MAP3K1 MLL2 MLL3 NOTCH3 NOTCH4 PDCD1 RNF43 \ + -st sample \ + -vt Consequence \ + -f tissue:intestine \ + -dpi 200 +``` +```python +# Python +import gget +gget.cbio_plot( + ['msk_impact_2017'], + ['AKT1', 'ALK', 'FLT4', 'MAP3K1', 'MLL2', 'MLL3', 'NOTCH3', 'NOTCH4', 'PDCD1', 'RNF43'], + stratification='sample', + variation_type='Consequence', + filter=('tissue', 'intestine'), + dpi=200, +) +``` + +→ Guarda un mapa de calor de tipos de mutaciones para los genes especificados en el estudio especificado, filtrado por tejido, en ./gget_cbio_figures/Heatmap_sample_intestine.png. + +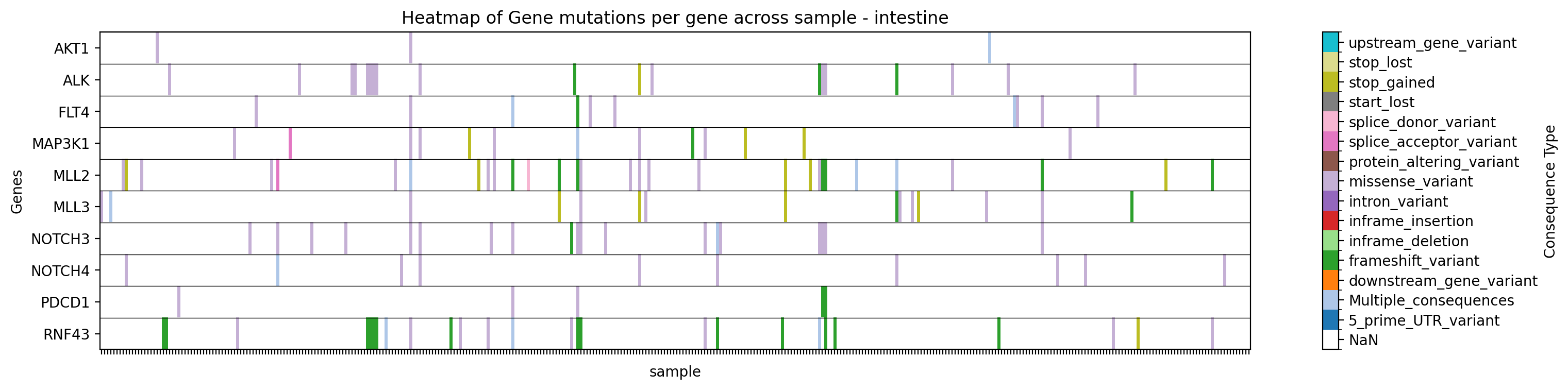 + +
+ +**Graficar un mapa de calor con un título y nombre de archivo personalizados::** +```bash +gget cbio plot \ + -s msk_impact_2017 \ + -g AKT1 ALK FLT4 MAP3K1 MLL2 MLL3 NOTCH3 NOTCH4 PDCD1 RNF43 \ + -st sample \ + -vt Consequence \ + -f tissue:intestine \ + -dpi 200 \ + -t "Intestinal Mutations" \ + -fn intestinal_mutations.png +``` +```python +# Python +import gget +gget.cbio_plot( + ['msk_impact_2017'], + ['AKT1', 'ALK', 'FLT4', 'MAP3K1', 'MLL2', 'MLL3', 'NOTCH3', 'NOTCH4', 'PDCD1', 'RNF43'], + stratification='sample', + variation_type='Consequence', + filter=('tissue', 'intestine'), + dpi=200, + figure_title='Intestinal Mutations', + figure_filename='intestinal_mutations.png' +) +``` + +→ Guarda un mapa de calor de los tipos de mutaciones para los genes especificados en el estudio especificado, filtrado por tejido, con el título "Mutaciones intestinales" en ./gget_cbio_figures/intestinal_mutations.png. + +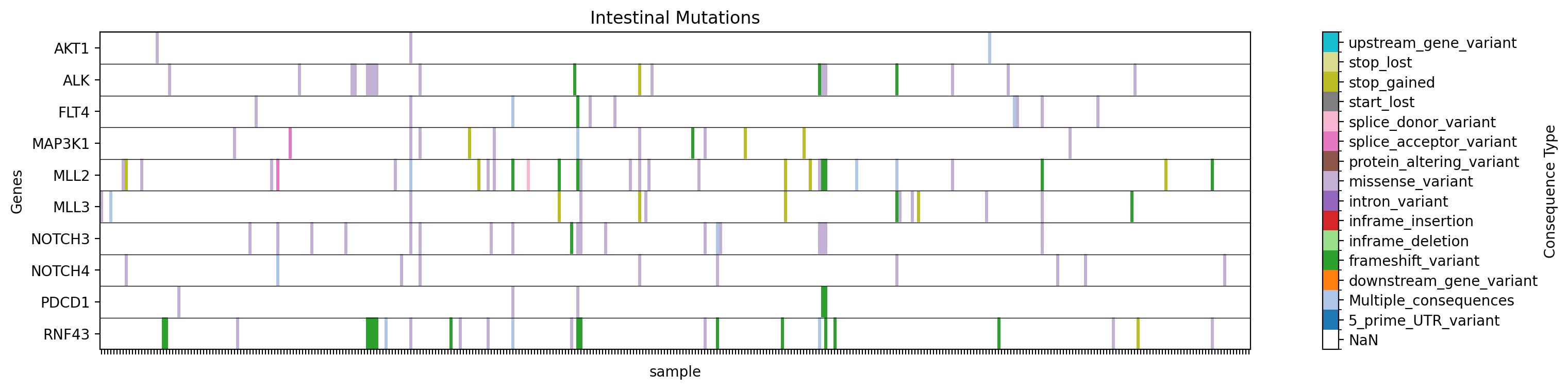 + +#### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget cbio` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012 May;2(5):401-4. doi: [10.1158/2159-8290.CD-12-0095](https://doi.org/10.1158/2159-8290.cd-12-0095). Erratum in: Cancer Discov. 2012 Oct;2(10):960. PMID: 22588877; PMCID: PMC3956037. + +- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013 Apr 2;6(269):pl1. doi: [10.1126/scisignal.2004088](https://doi.org/10.1126/scisignal.2004088). PMID: 23550210; PMCID: PMC4160307. + +- de Bruijn I, Kundra R, Mastrogiacomo B, Tran TN, Sikina L, Mazor T, Li X, Ochoa A, Zhao G, Lai B, Abeshouse A, Baiceanu D, Ciftci E, Dogrusoz U, Dufilie A, Erkoc Z, Garcia Lara E, Fu Z, Gross B, Haynes C, Heath A, Higgins D, Jagannathan P, Kalletla K, Kumari P, Lindsay J, Lisman A, Leenknegt B, Lukasse P, Madela D, Madupuri R, van Nierop P, Plantalech O, Quach J, Resnick AC, Rodenburg SYA, Satravada BA, Schaeffer F, Sheridan R, Singh J, Sirohi R, Sumer SO, van Hagen S, Wang A, Wilson M, Zhang H, Zhu K, Rusk N, Brown S, Lavery JA, Panageas KS, Rudolph JE, LeNoue-Newton ML, Warner JL, Guo X, Hunter-Zinck H, Yu TV, Pilai S, Nichols C, Gardos SM, Philip J; AACR Project GENIE BPC Core Team, AACR Project GENIE Consortium; Kehl KL, Riely GJ, Schrag D, Lee J, Fiandalo MV, Sweeney SM, Pugh TJ, Sander C, Cerami E, Gao J, Schultz N. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023 Dec 1;83(23):3861-3867. doi: [10.1158/0008-5472.CAN-23-0816](https://doi.org/10.1158/0008-5472.CAN-23-0816). PMID: 37668528; PMCID: PMC10690089. + +- Please also cite the source of the data if you are using a publicly available dataset. + diff --git a/docs/src/es/cellxgene.md b/docs/src/es/cellxgene.md index a6727546..53fae6f3 100644 --- a/docs/src/es/cellxgene.md +++ b/docs/src/es/cellxgene.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Las banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget cellxgene 🍱 +# gget cellxgene 🍱 Query data de la base de datos [CZ CELLxGENE Discover](https://cellxgene.cziscience.com/) usando [CZ CELLxGENE Discover Census](https://github.com/chanzuckerberg/cellxgene-census). Produce: Un objeto AnnData que contiene la matriz de recuentos de genes y los metadatos de resultados de single cell RNA-seq de los tejidos/genes/etcetera previamente definidos. @@ -135,3 +135,10 @@ df → Produce solo los metadatos de los conjuntos de datos de ENSMUSG00000015405 (ACE2), los cuales corresponden a células pulmonares murinas. Ver también: [https://chanzuckerberg.github.io/cellxgene-census/notebooks/api_demo/census_gget_demo.html](https://chanzuckerberg.github.io/cellxgene-census/notebooks/api_demo/census_gget_demo.html) + +# Citar +Si utiliza `gget cellxgene` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Chanzuckerberg Initiative. (n.d.). CZ CELLxGENE Discover. Retrieved [insert date here], from [https://cellxgene.cziscience.com/](https://cellxgene.cziscience.com/) diff --git a/docs/src/es/cite.md b/docs/src/es/cite.md index 6552eb8d..638d23af 100644 --- a/docs/src/es/cite.md +++ b/docs/src/es/cite.md @@ -5,48 +5,83 @@ Si utiliza `gget` en una publicación, favor de citar: Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. https://doi.org/10.1093/bioinformatics/btac836 -- Si utiliza `gget alphafold`, favor de citar: - - Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2 +- Si utiliza `gget alphafold`, favor de citar también: + - Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [https://doi.org/10.1038/s41586-021-03819-2](https://doi.org/10.1038/s41586-021-03819-2) Y si aplica: - - Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021.10.04.463034; https://doi.org/10.1101/2021.10.04.463034 + - Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021.10.04.463034; [https://doi.org/10.1101/2021.10.04.463034](https://doi.org/10.1101/2021.10.04.463034) -- Si utiliza `gget archs4`, favor de citar: +- Si utiliza `gget archs4`, favor de citar también: - Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, Wang L, Silverstein MC, Ma’ayan A. Massive mining of publicly available RNA-seq data from human and mouse. Nature Communications 9. Article number: 1366 (2018), doi:10.1038/s41467-018-03751-6 - - Bray NL, Pimentel H, Melsted P and Pachter L, Near optimal probabilistic RNA-seq quantification, Nature Biotechnology 34, p 525--527 (2016). https://doi.org/10.1038/nbt.3519 + - Bray NL, Pimentel H, Melsted P and Pachter L, Near optimal probabilistic RNA-seq quantification, Nature Biotechnology 34, p 525--527 (2016). [https://doi.org/10.1038/nbt.3519](https://doi.org/10.1038/nbt.3519) -- Si utilizan `gget blast`, por favor también de citar: +- Si utiliza `gget bgee`, favor de citar también: + - Frederic B Bastian, Julien Roux, Anne Niknejad, Aurélie Comte, Sara S Fonseca Costa, Tarcisio Mendes de Farias, Sébastien Moretti, Gilles Parmentier, Valentine Rech de Laval, Marta Rosikiewicz, Julien Wollbrett, Amina Echchiki, Angélique Escoriza, Walid H Gharib, Mar Gonzales-Porta, Yohan Jarosz, Balazs Laurenczy, Philippe Moret, Emilie Person, Patrick Roelli, Komal Sanjeev, Mathieu Seppey, Marc Robinson-Rechavi (2021). The Bgee suite: integrated curated expression atlas and comparative transcriptomics in animals. Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D831–D847, [https://doi.org/10.1093/nar/gkaa793](https://doi.org/10.1093/nar/gkaa793) + +- Si utiliza `gget blast`, favor de citar también: - Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990 Oct 5;215(3):403-10. doi: 10.1016/S0022-2836(05)80360-2. PMID: 2231712. -- Si utiliza `gget blat`, favor de citar: +- Si utiliza `gget blat`, favor de citar también: - Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002 Apr;12(4):656-64. doi: 10.1101/gr.229202. PMID: 11932250; PMCID: PMC187518. -- Si utiliza `gget cellxgene`, favor de citar: - - Chanzuckerberg Initiative. (n.d.). CZ CELLxGENE Discover. Retrieved [insert date here], from https://cellxgene.cziscience.com/ +- Si utiliza `gget cbio`, favor de citar también: + - Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012 May;2(5):401-4. doi: [10.1158/2159-8290.CD-12-0095](https://doi.org/10.1158/2159-8290.cd-12-0095). Erratum in: Cancer Discov. 2012 Oct;2(10):960. PMID: 22588877; PMCID: PMC3956037. + + - Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013 Apr 2;6(269):pl1. doi: [10.1126/scisignal.2004088](https://doi.org/10.1126/scisignal.2004088). PMID: 23550210; PMCID: PMC4160307. + + - de Bruijn I, Kundra R, Mastrogiacomo B, Tran TN, Sikina L, Mazor T, Li X, Ochoa A, Zhao G, Lai B, Abeshouse A, Baiceanu D, Ciftci E, Dogrusoz U, Dufilie A, Erkoc Z, Garcia Lara E, Fu Z, Gross B, Haynes C, Heath A, Higgins D, Jagannathan P, Kalletla K, Kumari P, Lindsay J, Lisman A, Leenknegt B, Lukasse P, Madela D, Madupuri R, van Nierop P, Plantalech O, Quach J, Resnick AC, Rodenburg SYA, Satravada BA, Schaeffer F, Sheridan R, Singh J, Sirohi R, Sumer SO, van Hagen S, Wang A, Wilson M, Zhang H, Zhu K, Rusk N, Brown S, Lavery JA, Panageas KS, Rudolph JE, LeNoue-Newton ML, Warner JL, Guo X, Hunter-Zinck H, Yu TV, Pilai S, Nichols C, Gardos SM, Philip J; AACR Project GENIE BPC Core Team, AACR Project GENIE Consortium; Kehl KL, Riely GJ, Schrag D, Lee J, Fiandalo MV, Sweeney SM, Pugh TJ, Sander C, Cerami E, Gao J, Schultz N. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023 Dec 1;83(23):3861-3867. doi: [10.1158/0008-5472.CAN-23-0816](https://doi.org/10.1158/0008-5472.CAN-23-0816). PMID: 37668528; PMCID: PMC10690089. -- Si utiliza `gget diamond`, favor de citar: - - Buchfink, B., Xie, C. & Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015). https://doi.org/10.1038/nmeth.3176 + - Please also cite the source of the data if you are using a publicly available dataset. + +- Si utiliza `gget cellxgene`, favor de citar también: + - Chanzuckerberg Initiative. (n.d.). CZ CELLxGENE Discover. Retrieved [insert date here], from [https://cellxgene.cziscience.com/](https://cellxgene.cziscience.com/) + +- Si utiliza `gget cosmic`, favor de citar también: + - Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, Fish P, Harsha B, Hathaway C, Jupe SC, Kok CY, Noble K, Ponting L, Ramshaw CC, Rye CE, Speedy HE, Stefancsik R, Thompson SL, Wang S, Ward S, Campbell PJ, Forbes SA. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019 Jan 8;47(D1):D941-D947. doi: [10.1093/nar/gky1015](https://doi.org/10.1093/nar/gky1015). PMID: 30371878; PMCID: PMC6323903. + +- Si utiliza `gget diamond`, favor de citar también: + - Buchfink, B., Xie, C. & Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015). [https://doi.org/10.1038/nmeth.3176](https://doi.org/10.1038/nmeth.3176) -- Si utiliza `gget elm`, favor de citar: +- Si utiliza `gget elm`, favor de citar también: - Laura Luebbert, Chi Hoang, Manjeet Kumar, Lior Pachter, Fast and scalable querying of eukaryotic linear motifs with gget elm, Bioinformatics, 2024, btae095, [https://doi.org/10.1093/bioinformatics/btae095](https://doi.org/10.1093/bioinformatics/btae095) - Manjeet Kumar, Sushama Michael, Jesús Alvarado-Valverde, Bálint Mészáros, Hugo Sámano‐Sánchez, András Zeke, Laszlo Dobson, Tamas Lazar, Mihkel Örd, Anurag Nagpal, Nazanin Farahi, Melanie Käser, Ramya Kraleti, Norman E Davey, Rita Pancsa, Lucía B Chemes, Toby J Gibson, The Eukaryotic Linear Motif resource: 2022 release, Nucleic Acids Research, Volume 50, Issue D1, 7 January 2022, Pages D497–D508, [https://doi.org/10.1093/nar/gkab975](https://doi.org/10.1093/nar/gkab975) - -- Si utiliza `gget enrichr`, favor de citar: - - Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. -Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 128(14). +- Si utiliza `gget enrichr`, favor de citar también: + - Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 128(14). [https://doi.org/10.1186/1471-2105-14-128 ](https://doi.org/10.1186/1471-2105-14-128) - - Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. -Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 2016; gkw377. + - Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 2016; gkw377. doi: [10.1093/nar/gkw377](https://doi.org/10.1093/nar/gkw377) - - Xie Z, Bailey A, Kuleshov MV, Clarke DJB., Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, Jeon M, & Ma’ayan A. -Gene set knowledge discovery with Enrichr. Current Protocols, 1, e90. 2021. doi: 10.1002/cpz1.90. + - Xie Z, Bailey A, Kuleshov MV, Clarke DJB., Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, Jeon M, & Ma’ayan A. Gene set knowledge discovery with Enrichr. Current Protocols, 1, e90. 2021. doi: [10.1002/cpz1.90](https://doi.org/10.1002/cpz1.90). + + Si trabaja con conjuntos de datos no humanos/ratón, cite también: + - Kuleshov MV, Diaz JEL, Flamholz ZN, Keenan AB, Lachmann A, Wojciechowicz ML, Cagan RL, Ma'ayan A. modEnrichr: a suite of gene set enrichment analysis tools for model organisms. Nucleic Acids Res. 2019 Jul 2;47(W1):W183-W190. doi: [10.1093/nar/gkz347](https://doi.org/10.1093/nar/gkz347). PMID: 31069376; PMCID: PMC6602483. + +- Si utiliza `gget info`, favor de citar también: + - Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. + + - Sayers EW, Beck J, Bolton EE, Brister JR, Chan J, Comeau DC, Connor R, DiCuccio M, Farrell CM, Feldgarden M, Fine AM, Funk K, Hatcher E, Hoeppner M, Kane M, Kannan S, Katz KS, Kelly C, Klimke W, Kim S, Kimchi A, Landrum M, Lathrop S, Lu Z, Malheiro A, Marchler-Bauer A, Murphy TD, Phan L, Prasad AB, Pujar S, Sawyer A, Schmieder E, Schneider VA, Schoch CL, Sharma S, Thibaud-Nissen F, Trawick BW, Venkatapathi T, Wang J, Pruitt KD, Sherry ST. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2024 Jan 5;52(D1):D33-D43. doi: [10.1093/nar/gkad1044](https://doi.org/10.1093/nar/gkad1044). PMID: 37994677; PMCID: PMC10767890. + + - The UniProt Consortium , UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Research, Volume 51, Issue D1, 6 January 2023, Pages D523–D531, [https://doi.org/10.1093/nar/gkac1052](https://doi.org/10.1093/nar/gkac1052) + +- Si utiliza `gget muscle`, favor de citar también: + - Edgar RC (2021), MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping, bioRxiv 2021.06.20.449169. [https://doi.org/10.1101/2021.06.20.449169](https://doi.org/10.1101/2021.06.20.449169) + +- Si utiliza `gget opentargets`, favor de citar también: + - Ochoa D, Hercules A, Carmona M, Suveges D, Baker J, Malangone C, Lopez I, Miranda A, Cruz-Castillo C, Fumis L, Bernal-Llinares M, Tsukanov K, Cornu H, Tsirigos K, Razuvayevskaya O, Buniello A, Schwartzentruber J, Karim M, Ariano B, Martinez Osorio RE, Ferrer J, Ge X, Machlitt-Northen S, Gonzalez-Uriarte A, Saha S, Tirunagari S, Mehta C, Roldán-Romero JM, Horswell S, Young S, Ghoussaini M, Hulcoop DG, Dunham I, McDonagh EM. The next-generation Open Targets Platform: reimagined, redesigned, rebuilt. Nucleic Acids Res. 2023 Jan 6;51(D1):D1353-D1359. doi: [10.1093/nar/gkac1046](https://doi.org/10.1093/nar/gkac1046). PMID: 36399499; PMCID: PMC9825572. + +- Si utiliza `gget pdb`, favor de citar también: + - Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000 Jan 1;28(1):235-42. doi: [10.1093/nar/28.1.235](https://doi.org/10.1093/nar/28.1.235). PMID: 10592235; PMCID: PMC102472. + +- Si utiliza `gget ref` o `gget search`, favor de citar también: + - Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. + +- Si utiliza `gget seq`, favor de citar también: + - Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. + + - The UniProt Consortium , UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Research, Volume 51, Issue D1, 6 January 2023, Pages D523–D531, [https://doi.org/10.1093/nar/gkac1052](https://doi.org/10.1093/nar/gkac1052) -- Si utiliza `gget muscle`, favor de citar: - - Edgar RC (2021), MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping, bioRxiv 2021.06.20.449169. https://doi.org/10.1101/2021.06.20.449169. - ___ # Descargo de responsabilidad `gget` es tan preciso como la base de datos/servidores/APIs que utiliza. La exactitud o fiabilidad de los datos no es garantizada por ningún motivo. Los proveedores por ningún motivo seran responsables de (incluyendo, sin limite alguno) la calidad, ejecución, o comerciabilidad para cualquier propósito particular surgiendo del uso o la incapacidad de usar los datos. diff --git a/docs/src/es/cosmic.md b/docs/src/es/cosmic.md index e6360414..c3c5e71c 100644 --- a/docs/src/es/cosmic.md +++ b/docs/src/es/cosmic.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Las banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget cosmic 🪐 +# gget cosmic 🪐 Busque genes, mutaciones, etc. asociados con cánceres utilizando la base de datos [COSMIC](https://cancer.sanger.ac.uk/cosmic) (Catálogo de mutaciones somáticas en cáncer). Produce: JSON (línea de comandos) o marco de datos/CSV (Python) cuando `download_cosmic=False`. Cuando `download_cosmic=True`, descarga la base de datos solicitada en la carpeta especificada. @@ -97,3 +97,10 @@ gget cosmic --download_cosmic gget.cosmic(searchterm=None, download_cosmic=True) ``` → Descargue la base de datos sobre cáncer de COSMIC de la última versión de COSMIC. + +# Citar +Si utiliza `gget cosmic` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, Fish P, Harsha B, Hathaway C, Jupe SC, Kok CY, Noble K, Ponting L, Ramshaw CC, Rye CE, Speedy HE, Stefancsik R, Thompson SL, Wang S, Ward S, Campbell PJ, Forbes SA. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019 Jan 8;47(D1):D941-D947. doi: [10.1093/nar/gky1015](https://doi.org/10.1093/nar/gky1015). PMID: 30371878; PMCID: PMC6323903. diff --git a/docs/src/es/dependents.md b/docs/src/es/dependents.md new file mode 100644 index 00000000..c5d73000 --- /dev/null +++ b/docs/src/es/dependents.md @@ -0,0 +1,59 @@ +
+ Usuarios activos del sitio web de gget desde su creación: +
+ +[](https://github.com/lauraluebbert/lauraluebbert) + +# 🧑🤝🧑 Dependientes +Las siguientes aplicaciones se basan en *gget*: +- [Therapeutics Data Commons (TDC)](https://tdcommons.ai/) + Base de inteligencia artificial para la ciencia terapéutica ([código fuente](https://github.com/mims-harvard/TDC), [artículo en Nat Chem Bio](https://www.nature.com/articles/s41589-022-01131-2)) del laboratorio de Inteligencia Artificial para Medicina y Ciencia de Harvard. +- [BioDiscoveryAgent](https://github.com/snap-stanford/BioDiscoveryAgent) + BioDiscoveryAgent es un agente de IA basado en modelos de lenguaje para el diseño en bucle cerrado de experimentos de perturbación genética ([preprint](https://arxiv.org/abs/2405.17631)) del Proyecto de Análisis de Redes de Stanford. +- [DeepChopper](https://ylab-hi.github.io/DeepChopper/) + Modelos de lenguaje para identificar lecturas artificiales quiméricas en datos de secuenciación directa de ARN de NanoPore por el laboratorio de Yang en Northwestern. +- [BRAD](https://github.com/Jpickard1/BRAD) + Un chatbot impulsado por un modelo de lenguaje para bioinformática ([documentación](https://brad-bioinformatics-retrieval-augmented-data.readthedocs.io/en/latest/index.html), [página principal del proyecto](https://brad-bioinformatics-retrieval-augmented-data.readthedocs.io/_/downloads/en/latest/pdf/)). +- [scPRINT](https://www.jkobject.com/scPRINT/) + scPRINT es un modelo transformer grande diseñado para inferir redes génicas (conexiones entre genes que explican el perfil de expresión de la célula) a partir de datos de scRNAseq ([preprint](https://www.biorxiv.org/content/10.1101/2024.07.29.605556v1)). +- [AnoPrimer](https://sanjaynagi.github.io/AnoPrimer/landing-page.html) + AnoPrimer es un paquete de Python para el diseño de cebadores en *An. gambiae* y *An. funestus*, teniendo en cuenta la variación genética en especímenes de genomas completos secuenciados de la naturaleza en datos de malariagen. +- [AvaTaR](https://github.com/zou-group/avatar) + Optimización de Agentes de LLM para Recuperación de Conocimiento Asistida por Herramientas (NeurIPS 2024) por el laboratorio de James Zou en la Universidad de Stanford. +- [GRLDrugProp](https://github.com/Madscba/GRLDrugProp) + Aprendizaje de representación de grafos para modelar propiedades de fármacos. +- Implementación en Rust de gget: [https://github.com/noamteyssier/ggetrs](https://github.com/noamteyssier/ggetrs) +- [https://github.com/Superbio-ai/getbio](https://github.com/Superbio-ai/getbio) +- [https://github.com/yonniejon/AchillesPrediction](https://github.com/yonniejon/AchillesPrediction) +- [https://github.com/ELELAB/cancermuts](https://github.com/ELELAB/cancermuts) +- [https://github.com/Benoitdw/SNPrimer](https://github.com/Benoitdw/SNPrimer) +- [https://github.com/louisjoecodes/a16z-hackathon-project](https://github.com/louisjoecodes/a16z-hackathon-project) +- [https://github.com/EvX57/BACE1-Drug-Discovery](https://github.com/EvX57/BACE1-Drug-Discovery) +- [https://github.com/vecerkovakaterina/hidden-genes-msc](https://github.com/vecerkovakaterina/hidden-genes-msc) +- [https://github.com/vecerkovakaterina/llm_bioinfo_agent](https://github.com/vecerkovakaterina/llm_bioinfo_agent) +- [https://github.com/greedjar74/upstage_AI_Lab](https://github.com/greedjar74/upstage_AI_Lab) +- [https://github.com/alphavector/all](https://github.com/alphavector/all) + +Ver también: [https://github.com/pachterlab/gget/network/dependents](https://github.com/pachterlab/gget/network/dependents) + +# 📃 Publicaciones destacadas +- David Bradley et al., [The fitness cost of spurious phosphorylation.](https://doi.org/10.1038/s44318-024-00200-7) *The EMBO Journal* (2024). DOI: 10.1038/s44318-024-00200-7 +- Mikael Nilsson et al., [Resolving thyroid lineage cell trajectories merging into a dual endocrine gland in mammals.](https://doi.org/10.21203/rs.3.rs-5278325/v1) *Nature Portfolio (en revisión)* (2024). DOI: 10.21203/rs.3.rs-5278325/v1 +- Avasthi P et al., [Repeat expansions associated with human disease are present in diverse organisms.](https://doi.org/10.57844/arcadia-e367-8b55) *Arcadia* (2024). DOI: 10.57844/arcadia-e367-8b55 +- Ibrahim Al Rayyes et al., [Single-Cell Transcriptomics Reveals the Molecular Logic Underlying Ca2+ Signaling Diversity in Human and Mouse Brain.](https://doi.org/10.1101/2024.04.26.591400) *bioRxiv* (2024). DOI: 10.1101/2024.04.26.591400 +- David R. Blair & Neil Risch. [Dissecting the Reduced Penetrance of Putative Loss-of-Function Variants in Population-Scale Biobanks.](https://doi.org/10.1101/2024.09.23.24314008) *medRxiv* (2024). DOI: 10.1101/2024.09.23.24314008 +- Shanmugampillai Jeyarajaguru Kabilan et al., [Molecular modelling approaches for the identification of potent Sodium-Glucose Cotransporter 2 inhibitors from Boerhavia diffusa for the potential treatment of chronic kidney disease.](https://doi.org/10.21203/rs.3.rs-4520611/v1) *Journal of Computer-Aided Molecular Design (en revisión)* (2024). DOI: 10.21203/rs.3.rs-4520611/v1 +- Joseph M Rich et al., [The impact of package selection and versioning on single-cell RNA-seq analysis.](https://pmc.ncbi.nlm.nih.gov/articles/PMC11014608/#:~:text=10.1101/2024.04.04.588111) *bioRxiv* (2024). DOI: 10.1101/2024.04.04.588111 +- Sanjay C. Nagi et al., [AnoPrimer: Primer Design in malaria vectors informed by range-wide genomic variation.](https://wellcomeopenresearch.org/articles/9-255/v1) *Wellcome Open Research* (2024). +- Yasmin Makki Mohialden et al., [A survey of the most recent Python packages for use in biology.](http://dx.doi.org/10.48047/NQ.2023.21.2.NQ23029) *NeuroQuantology* (2023). DOI: 10.48047/NQ.2023.21.2.NQ23029 +- Kimberly Siletti et al., [Transcriptomic diversity of cell types across the adult human brain.](https://doi.org/10.1126/science.add7046) *Science* (2023). DOI: 10.1126/science.add7046 +- Beatriz Beamud et al., [Genetic determinants of host tropism in Klebsiella phages.](https://doi.org/10.1016/j.celrep.2023.112048) *Cell Reports* (2023). DOI: 10.1016/j.celrep.2023.112048 +- Nicola A. Kearns et al., [Generation and molecular characterization of human pluripotent stem cell-derived pharyngeal foregut endoderm.](https://doi.org/10.1016/j.devcel.2023.08.024) *Cell Reports* (2023). DOI: 10.1016/j.devcel.2023.08.024 +- Jonathan Rosenski et al., [Predicting gene knockout effects from expression data.](https://link.springer.com/article/10.1186/s12920-023-01446-6) *BMC Medical Genomics* (2023). DOI: 10.1186/s12920-023-01446-6 +- Peter Overby et al., [Pharmacological or genetic inhibition of Scn9a protects beta-cells while reducing insulin secretion in type 1 diabetes.](https://doi.org/10.1101/2023.06.11.544521) *bioRxiv* (2023). DOI: 10.1101/2023.06.11.544521 +- Mingze Dong et al., [Deep identifiable modeling of single-cell atlases enables zero-shot query of cellular states.](https://doi.org/10.1101/2023.11.11.566161) *bioRxiv* (2023). DOI: 10.1101/2023.11.11.566161 + +# 📰 Noticias +- Documental corto sobre *gget*: [https://youtu.be/cVR0k6Mt97o](https://youtu.be/cVR0k6Mt97o) +- Episodio de podcast para el Prototype Fund Public Interest Podcast sobre la importancia del software de código abierto y su papel en la investigación académica (en alemán): [https://public-interest-podcast.podigee.io/33-pips4e4](https://public-interest-podcast.podigee.io/33-pips4e4) +- Anuncio del Prototype Fund: [https://prototypefund.de/project/gget-genomische-datenbanken](https://prototypefund.de/project/gget-genomische-datenbanken/) diff --git a/docs/src/es/diamond.md b/docs/src/es/diamond.md index 2e064d10..e66593f7 100644 --- a/docs/src/es/diamond.md +++ b/docs/src/es/diamond.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Las banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget diamond 💎 +# gget diamond 💎 Alinee múltiples proteínas o secuencias de ADN traducidas usando [DIAMOND](https://www.nature.com/articles/nmeth.3176) (DIAMOND es similar a BLAST, pero este es un cálculo local). Produce: Resultados en formato JSON (Terminal) o Dataframe/CSV (Python). @@ -60,3 +60,10 @@ gget.diamond(["GGETISAWESQME", "ELVISISALIVE", "LQVEFRANKLIN", "PACHTERLABRQCKS" #### [Màs ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget diamond` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Buchfink, B., Xie, C. & Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015). [https://doi.org/10.1038/nmeth.3176](https://doi.org/10.1038/nmeth.3176) diff --git a/docs/src/es/elm.md b/docs/src/es/elm.md index 2a1fb54d..ebfa5fa4 100644 --- a/docs/src/es/elm.md +++ b/docs/src/es/elm.md @@ -1,14 +1,10 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget elm 🎭 +# gget elm 🎭 Prediga localmente motivos lineales eucarióticos (ELMs) a partir de una secuencia de aminoácidos o UniProt Acc utilizando datos de la [base de datos ELM](http://elm.eu.org/). Produce: Resultados en formato JSON (Terminal) o Dataframe/CSV (Python). Este módulo devuelve dos tipos de resultados (ver ejemplos). **Los datos de ELM se pueden descargar y distribuir para uso no comercial de acuerdo con el [acuerdo de licencia de software de ELM](http://elm.eu.org/media/Elm_academic_license.pdf).** -Si utiliza `gget elm` en una publicación, favor de citar: -- Laura Luebbert, Chi Hoang, Manjeet Kumar, Lior Pachter, Fast and scalable querying of eukaryotic linear motifs with gget elm, _Bioinformatics_, 2024, btae095, [https://doi.org/10.1093/bioinformatics/btae095](https://doi.org/10.1093/bioinformatics/btae095) -- Manjeet Kumar, _et al._, The Eukaryotic Linear Motif resource: 2022 release, _Nucleic Acids Research_, Volume 50, Issue D1, 7 January 2022, Pages D497–D508, [https://doi.org/10.1093/nar/gkab975](https://doi.org/10.1093/nar/gkab975) - Antes de usar `gget elm` por primera vez, ejecute `gget setup elm` / `gget.setup("elm")` una vez (consulte también [`gget setup`](setup.md)). **Parámetro posicional** @@ -90,3 +86,10 @@ regex_df: (Los motivos que aparecen en muchas especies diferentes pueden parecer repetidos, pero todas las filas deben ser únicas.) #### [Màs ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget elm` en una publicación, favor de citar los siguientes artículos: +- Laura Luebbert, Chi Hoang, Manjeet Kumar, Lior Pachter, Fast and scalable querying of eukaryotic linear motifs with gget elm, _Bioinformatics_, 2024, btae095, [https://doi.org/10.1093/bioinformatics/btae095](https://doi.org/10.1093/bioinformatics/btae095) + +- Manjeet Kumar, Sushama Michael, Jesús Alvarado-Valverde, Bálint Mészáros, Hugo Sámano‐Sánchez, András Zeke, Laszlo Dobson, Tamas Lazar, Mihkel Örd, Anurag Nagpal, Nazanin Farahi, Melanie Käser, Ramya Kraleti, Norman E Davey, Rita Pancsa, Lucía B Chemes, Toby J Gibson, The Eukaryotic Linear Motif resource: 2022 release, Nucleic Acids Research, Volume 50, Issue D1, 7 January 2022, Pages D497–D508, [https://doi.org/10.1093/nar/gkab975](https://doi.org/10.1093/nar/gkab975) + diff --git a/docs/src/es/enrichr.md b/docs/src/es/enrichr.md index 8ab83762..8fa07b34 100644 --- a/docs/src/es/enrichr.md +++ b/docs/src/es/enrichr.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Las banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget enrichr 💰 +# gget enrichr 💰 Realice un análisis de enriquecimiento de una lista de genes utilizando [Enrichr](https://maayanlab.cloud/Enrichr/). Produce: Resultados en formato JSON (Terminal) o Dataframe/CSV (Python). @@ -20,6 +20,19 @@ Admite cualquier base de datos enumerada [aquí](https://maayanlab.cloud/Enrichr 'kinase_interactions' (KEA_2015) **Parámetros opcionales** +`-s` `--species` +Especies a utilizar como referencia para el análisis de enriquecimiento. (Por defecto: human) +Opciones: + +| Species | Database list | +|----------|-------------------------------------------------------------------| +| `human` | [Enrichr](https://maayanlab.cloud/Enrichr/#libraries) | +| `mouse` | [Equivalente al humano](https://maayanlab.cloud/Enrichr/#libraries) | +| `fly` | [FlyEnrichr](https://maayanlab.cloud/FlyEnrichr/#stats) | +| `yeast` | [YeastEnrichr](https://maayanlab.cloud/YeastEnrichr/#stats) | +| `worm` | [WormEnrichr](https://maayanlab.cloud/WormEnrichr/#stats) | +| `fish` | [FishEnrichr](https://maayanlab.cloud/FishEnrichr/#stats) | + `-bkg_l` `--background_list` Lista de nombres cortos (símbolos) de genes de 'background' (de fondo/control), p. NSUN3 POLRMT NLRX1. Alternativamente: usa la bandera `--ensembl_background` para ingresar IDs tipo Ensembl. @@ -205,3 +218,17 @@ df |> ``` #### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget enrichr` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 128(14). [https://doi.org/10.1186/1471-2105-14-128 ](https://doi.org/10.1186/1471-2105-14-128) + +- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 2016; gkw377. doi: [10.1093/nar/gkw377](https://doi.org/10.1093/nar/gkw377) + +- Xie Z, Bailey A, Kuleshov MV, Clarke DJB., Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, Jeon M, & Ma’ayan A. Gene set knowledge discovery with Enrichr. Current Protocols, 1, e90. 2021. doi: [10.1002/cpz1.90](https://doi.org/10.1002/cpz1.90). + +Si trabaja con conjuntos de datos no humanos/ratón, cite también: +- Kuleshov MV, Diaz JEL, Flamholz ZN, Keenan AB, Lachmann A, Wojciechowicz ML, Cagan RL, Ma'ayan A. modEnrichr: a suite of gene set enrichment analysis tools for model organisms. Nucleic Acids Res. 2019 Jul 2;47(W1):W183-W190. doi: [10.1093/nar/gkz347](https://doi.org/10.1093/nar/gkz347). PMID: 31069376; PMCID: PMC6602483. diff --git a/docs/src/es/gpt.md b/docs/src/es/gpt.md index a04c8e8c..9ec9087e 100644 --- a/docs/src/es/gpt.md +++ b/docs/src/es/gpt.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget gpt 💬 +# gget gpt 💬 Genera texto en lenguaje natural basado en mensaje de entrada. `gget gpt` use la API 'openai.ChatCompletion.create' de [OpenAI](https://openai.com/). Este módulo, incluido su código, documentación y pruebas unitarias, fue escrito en parte por Chat-GTP3 de OpenAI. diff --git a/docs/src/es/info.md b/docs/src/es/info.md index 8141aac8..72cb1fcf 100644 --- a/docs/src/es/info.md +++ b/docs/src/es/info.md @@ -1,11 +1,12 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget info 💡 +# gget info 💡 Obtenga información detallada sobre genes y transcripciones de [Ensembl](https://www.ensembl.org/), [UniProt](https://www.uniprot.org/) y [NCBI](https://www.ncbi.nlm.nih.gov/) utilizando sus IDs del tipo Ensembl. Regresa: Resultados en formato JSON (Terminal) o Dataframe/CSV (Python). **Parámetro posicional** `ens_ids` Uno o más ID del tipo Ensembl. +NOTA: Proporcionar una lista de más de 1000 ID de Ensembl a la vez puede provocar un error del servidor (para procesar más de 1000 ID, divida la lista de ID en fragmentos de 1000 ID y ejecútelos por separado). **Parámetros optionales** `-o` `--out` @@ -53,3 +54,14 @@ gget.info(["ENSG00000034713", "ENSG00000104853", "ENSG00000170296"]) | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | ... | #### [More examples](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget info` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. + +- Sayers EW, Beck J, Bolton EE, Brister JR, Chan J, Comeau DC, Connor R, DiCuccio M, Farrell CM, Feldgarden M, Fine AM, Funk K, Hatcher E, Hoeppner M, Kane M, Kannan S, Katz KS, Kelly C, Klimke W, Kim S, Kimchi A, Landrum M, Lathrop S, Lu Z, Malheiro A, Marchler-Bauer A, Murphy TD, Phan L, Prasad AB, Pujar S, Sawyer A, Schmieder E, Schneider VA, Schoch CL, Sharma S, Thibaud-Nissen F, Trawick BW, Venkatapathi T, Wang J, Pruitt KD, Sherry ST. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2024 Jan 5;52(D1):D33-D43. doi: [10.1093/nar/gkad1044](https://doi.org/10.1093/nar/gkad1044). PMID: 37994677; PMCID: PMC10767890. + +- The UniProt Consortium , UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Research, Volume 51, Issue D1, 6 January 2023, Pages D523–D531, [https://doi.org/10.1093/nar/gkac1052](https://doi.org/10.1093/nar/gkac1052) diff --git a/docs/src/es/installation.md b/docs/src/es/installation.md index a0e7be4f..b4777cc4 100644 --- a/docs/src/es/installation.md +++ b/docs/src/es/installation.md @@ -1,6 +1,6 @@ [](https://pypi.org/project/gget) [](https://anaconda.org/bioconda/gget) -## Instalación +# Instalación ```bash pip install --upgrade gget ``` diff --git a/docs/src/es/introduction.md b/docs/src/es/introduction.md index 06fb2aca..05a22649 100644 --- a/docs/src/es/introduction.md +++ b/docs/src/es/introduction.md @@ -20,24 +20,41 @@ Las bases de datos consultadas por `gget` se actualizan continuamente, lo que a [Solicitar una nueva función
](https://github.com/pachterlab/gget/issues/new/choose)
+# Módulos gget -[
 ](alphafold.md)
-[
](alphafold.md)
-[ ](archs4.md)
-[
](archs4.md)
-[ ](blast.md)
+Estos son los módulos principales de `gget`. Haga clic en cualquier módulo para acceder a la documentación detallada.
-[
](blast.md)
+Estos son los módulos principales de `gget`. Haga clic en cualquier módulo para acceder a la documentación detallada.
-[ ](blat.md)
-[
](blat.md)
-[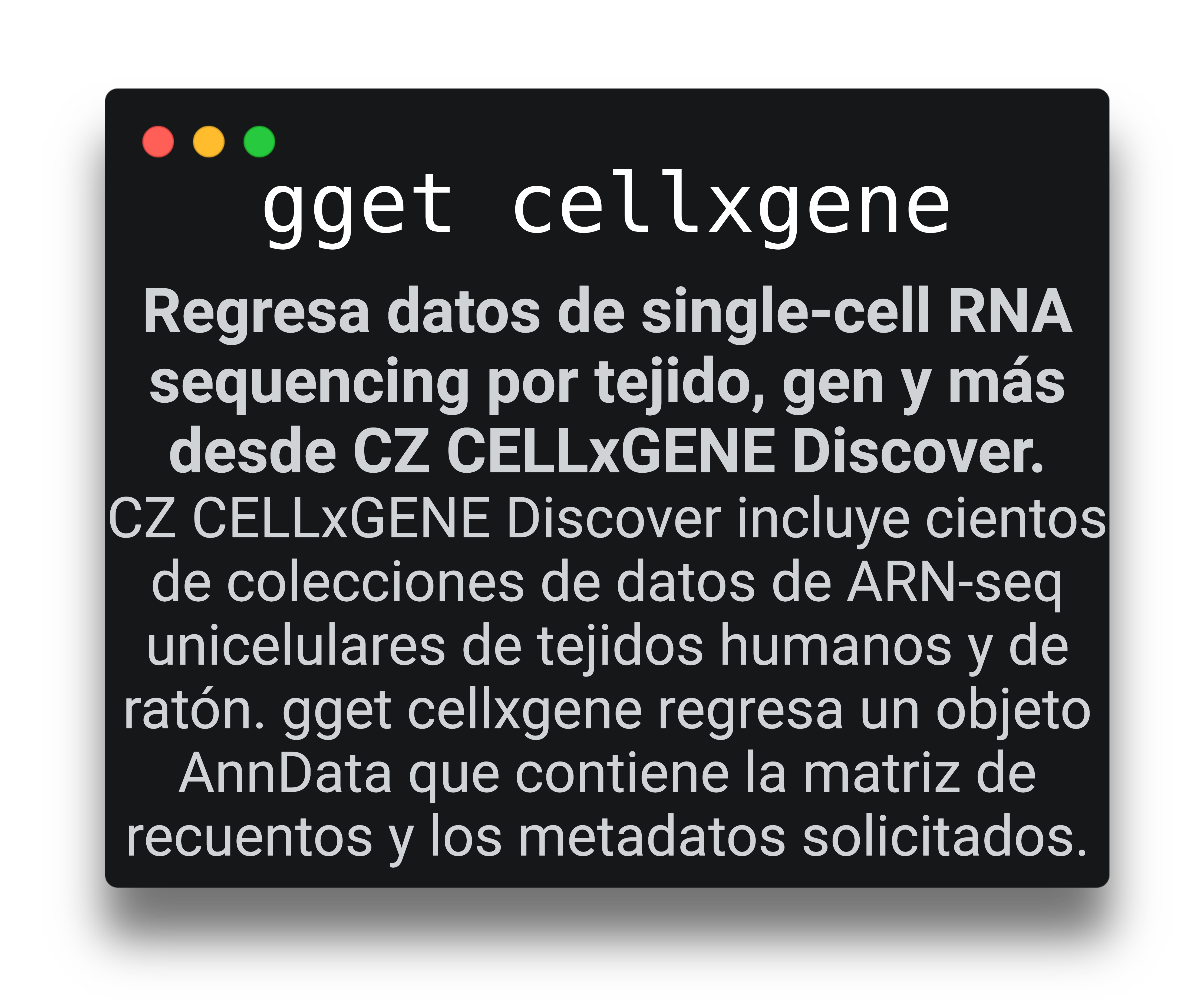 ](cellxgene.md)
-[
](cellxgene.md)
-[ ](enrichr.md)
-
-[
](enrichr.md)
-
-[ ](info.md)
-[
](info.md)
-[ ](muscle.md)
-[
](muscle.md)
-[ ](pdb.md)
-
-[
](pdb.md)
-
-[ ](ref.md)
-[
](ref.md)
-[ ](search.md)
-[
](search.md)
-[ ](seq.md)
-
-### [Más tutoriales](https://github.com/pachterlab/gget_examples)
+
](seq.md)
-
-### [Más tutoriales](https://github.com/pachterlab/gget_examples)
+| gget alphafold Predecir la estructura 3D de una proteína a partir de una secuencia de aminoácidos. |
+ gget archs4 ¿Cuál es la expresión de mi gen en el tejido X? |
+ gget bgee Encontrar todos los ortólogos de un gen. |
+ gget blast Realizar un BLAST de una secuencia de nucleótidos o aminoácidos. |
+
| gget blat Encontrar la ubicación genómica de una secuencia de nucleótidos o aminoácidos. |
+ gget cbio Explorar la expresión de un gen en los cánceres especificados. |
+ gget cellxgene Obtener matrices de conteo de ARN de células individuales listas para usar para ciertos tejidos/enfermedades/etc. |
+ gget cosmic Buscar genes, mutaciones y otros factores asociados con ciertos cánceres. |
+
| gget diamond Alinear secuencias de aminoácidos a una referencia. |
+ gget elm Encontrar dominios y funciones de interacción de proteínas en una secuencia de aminoácidos. |
+ gget enrichr Verificar si una lista de genes está asociada con un tipo celular específico/ vía/ enfermedad/ etc. |
+ gget info Recuperar toda la información asociada con un ID de Ensembl. |
+
| gget muscle Alinear múltiples secuencias de nucleótidos o aminoácidos entre sí. |
+ gget mutate Mutar secuencias de nucleótidos según mutaciones específicas. |
+ gget opentargets Explorar qué enfermedades y medicamentos están asociados con un gen. |
+ gget pdb Recuperar datos de la Base de Datos de Proteínas (PDB) según un ID de PDB. |
+
| gget ref Obtener genomas de referencia de Ensembl. |
+ gget search Encontrar IDs de Ensembl asociados con la palabra de búsqueda especificada. |
+ gget seq Recuperar la secuencia de nucleótidos o aminoácidos de un gen. |
+
@@ -51,3 +68,6 @@ Gracias a [Victor Garcia-Ruiz](https://github.com/victorg775) y [Anna Karen Orta
+ + + diff --git a/docs/src/es/muscle.md b/docs/src/es/muscle.md index 68a97345..539bc63e 100644 --- a/docs/src/es/muscle.md +++ b/docs/src/es/muscle.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget muscle 🦾 +# gget muscle 🦾 Alinea múltiples secuencias de nucleótidos o aminoácidos usando el algoritmo [Muscle5](https://www.drive5.com/muscle/). Regresa: Salida estándar (STDOUT) en formato ClustalW o archivo de tipo 'aligned FASTA' (.afa). @@ -56,3 +56,10 @@ alv.view(msa) ``` #### [More examples](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget muscle` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Edgar RC (2021), MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping, bioRxiv 2021.06.20.449169. [https://doi.org/10.1101/2021.06.20.449169](https://doi.org/10.1101/2021.06.20.449169) diff --git a/docs/src/es/mutate.md b/docs/src/es/mutate.md index af67d677..3b93056c 100644 --- a/docs/src/es/mutate.md +++ b/docs/src/es/mutate.md @@ -1,69 +1,124 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget mutate 🧟 +# gget mutate 🧟 Recibe secuencias de nucleótidos y mutaciones (en [anotación de mutación estándar](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1867422/)) y devuelve versiones mutadas de las secuencias según las mutaciones proporcionadas. -Formato de devolución: Guarda las secuencias mutadas en formato FASTA (o devuelve una lista que contiene las secuencias mutadas si `out=None`). +Resultado: Guarda las secuencias mutadas en formato FASTA (o devuelve una lista que contiene las secuencias mutadas si `out=None`). Este módulo fue coescrito por [Joseph Rich](https://github.com/josephrich98). -**Parámetro posicional** -`sequences` -Ruta al archivo FASTA que contiene las secuencias a mutar, por ejemplo, 'path/to/seqs.fa'. -Los identificadores de secuencia que siguen al carácter '>' deben corresponder a los identificadores en la columna seq_ID de mutations. -NOTA: Solo se utilizará la cadena que sigue al '>' hasta el primer espacio o punto como identificador de secuencia. -> Se ignorarán los números de versión de los IDs de Ensembl. +**Argumento posicional** +`sequences` +Ruta al archivo FASTA que contiene las secuencias a ser mutadas, por ejemplo, 'path/to/seqs.fa'. +Los identificadores de las secuencias que siguen al carácter '>' deben corresponder a los identificadores en la columna seq_ID de `mutations`. -Ejemplo de formato del archivo FASTA: +Formato de ejemplo del archivo FASTA: ``` ->seq1 (o ENSG00000106443) +>seq1 (or ENSG00000106443) ACTGCGATAGACT >seq2 AGATCGCTAG ``` -Alternativamente: Proporcione secuencia(s) como una cadena o lista, por ejemplo, 'AGCTAGCT'. +Alternativamente: Secuencia(s) de entrada como una cadena o lista, por ejemplo, 'AGCTAGCT'. -**Otros parámetros requeridos** +NOTA: Solo se utilizarán las letras hasta el primer espacio o punto como identificadores de secuencias; se ignorarán los números de versión de los IDs de Ensembl. +NOTA: Cuando la entrada `sequences` es un archivo fasta de genoma, consulte también el argumento `gtf` a continuación. + +**Argumentos requeridos** `-m` `--mutations` -Ruta al archivo csv o tsv (por ejemplo, 'path/to/mutations.csv') o marco de datos (objeto DataFrame) que contiene información sobre las mutaciones en el siguiente formato (la columna 'notas' no es necesaria): +Ruta al archivo csv o tsv (por ejemplo, 'path/to/mutations.csv') o marco de datos (objeto DataFrame) que contiene información sobre las mutaciones en el siguiente formato (las columnas 'notes' y 'mut_ID' son opcionales): -| mutation | mut_ID | seq_ID | notas | -|------------------|--------|--------|-| -| c.2C>T | mut1 | seq1 | -> Aplicar la mutación 1 a la secuencia 1 | -| c.9_13inv | mut2 | seq2 | -> Aplicar la mutación 2 a la secuencia 2 | -| c.9_13inv | mut2 | seq4 | -> Aplicar la mutación 2 a la secuencia 4 | -| c.9_13delinsAAT | mut3 | seq4 | -> Aplicar la mutación 3 a la secuencia 4 | -| ... | ... | ... | | +| mutation | mut_ID | seq_ID | notes | +|------------------|--------|--------|-------| +| c.2C>T | mut1 | seq1 | -> Aplicar mutación 1 a la secuencia 1 | +| c.9_13inv | mut2 | seq2 | -> Aplicar mutación 2 a la secuencia 2 | +| c.9_13inv | mut2 | seq4 | -> Aplicar mutación 2 a la secuencia 4 | +| c.9_13delinsAAT | mut3 | seq4 | -> Aplicar mutación 3 a la secuencia 4 | +| ... | ... | ... | | -'mutation' = Columna que contiene las mutaciones a realizar, escritas en anotación de mutación estándar -'mut_ID' = Columna que contiene el identificador de cada mutación -'seq_ID' = Columna que contiene los identificadores de las secuencias a mutar (deben corresponder a la cadena que sigue al carácter '>' en el archivo FASTA de 'sequences'; NO incluir espacios ni puntos) +'mutation' = Columna que contiene las mutaciones a realizar escritas en la anotación estándar de mutaciones +'mut_ID' = Columna que contiene el identificador para cada mutación +'seq_ID' = Columna que contiene los identificadores de las secuencias a ser mutadas (deben corresponder a la cadena que sigue al carácter '>' en el archivo FASTA 'sequences'; NO incluya espacios ni puntos) Alternativamente: Mutación(es) de entrada como una cadena o lista, por ejemplo, 'c.2C>T'. Si se proporciona una lista, el número de mutaciones debe ser igual al número de secuencias de entrada. -Para uso desde el terminal (bash): Encierre las anotaciones de mutación individuales entre comillas para evitar errores. -**Parámetros opcionales** -`-k` `--k` -Longitud de las secuencias que flanquean la mutación. Por defecto: 30. -Si k > longitud total de la secuencia, se mantendrá toda la secuencia. +Para usar desde la terminal (bash): Enciérrale las anotaciones de mutación individuales entre comillas para evitar errores de análisis. +**Argumentos opcionales relacionados con la entrada** `-mc` `--mut_column` -Nombre de la columna que contiene las mutaciones a realizar en `mutations`. Por defecto: 'mutation'. +Nombre de la columna que contiene las mutaciones a realizar en `mutations`. Predeterminado: 'mutation'. + +`-sic` `--seq_id_column` +Nombre de la columna que contiene los ID de las secuencias a ser mutadas en `mutations`. Predeterminado: 'seq_ID'. `-mic` `--mut_id_column` -Nombre de la columna que contiene los IDs de cada mutación en `mutations`. Por defecto: 'mut_ID'. +Nombre de la columna que contiene los IDs de cada mutación en `mutations`. Predeterminado: Igual que `mut_column`. -`-sic` `--seq_id_column` -Nombre de la columna que contiene los IDs de las secuencias a mutar en `mutations`. Por defecto: 'seq_ID'. +`-gtf` `--gtf` +Ruta a un archivo .gtf. Al proporcionar un archivo fasta de genoma como entrada para 'sequences', puede proporcionar un archivo .gtf aquí y las secuencias de entrada se definirán de acuerdo con los límites de los transcritos, por ejemplo, 'path/to/genome_annotation.gtf'. Predeterminado: Ninguno + +`-gtic` `--gtf_transcript_id_column` +Nombre de la columna en el archivo de entrada `mutations` que contiene el ID del transcrito. En este caso, la columna `seq_id_column` debe contener el número de cromosoma. +Requerido cuando se proporciona `gtf`. Predeterminado: Ninguno + +**Argumentos opcionales para la generación/filtrado de secuencias mutantes** +`-k` `--k` +Longitud de las secuencias que flanquean la mutación. Predeterminado: 30. +Si k > longitud total de la secuencia, se mantendrá toda la secuencia. + +`-msl` `--min_seq_len` +Longitud mínima de la secuencia de salida mutante, por ejemplo, 100. Las secuencias mutantes más pequeñas que esto serán descartadas. Predeterminado: Ninguno + +`-ma` `--max_ambiguous` +Número máximo de caracteres 'N' (o 'n') permitidos en la secuencia de salida, por ejemplo, 10. Predeterminado: Ninguno (no se aplicará filtro de caracteres ambiguos) + +**Banderas opcionales para la generación/filtrado de secuencias mutantes** +`-ofr` `--optimize_flanking_regions` +Elimina nucleótidos de cualquiera de los extremos de la secuencia mutante para asegurar (cuando sea posible) que la secuencia mutante no contenga ningún k-mer que también se encuentre en la secuencia de tipo salvaje/entrada. +`-rswk` `--remove_seqs_with_wt_kmers` +Elimina las secuencias de salida donde al menos un k-mer también está presente en la secuencia de tipo salvaje/entrada en la misma región. +Cuando se utiliza con `--optimize_flanking_regions`, solo se eliminarán las secuencias para las cuales un k-mer de tipo salvaje aún está presente después de la optimización. + +`-mio` `--merge_identical_off` +No fusionar secuencias mutantes idénticas en la salida (por defecto, las secuencias idénticas se fusionarán concatenando los encabezados de secuencia para todas las secuencias idénticas). + +**Argumentos opcionales para generar salida adicional** +Esta salida se activa utilizando la bandera `--update_df` y se almacenará en una copia del DataFrame `mutations`. + +`-udf_o` `--update_df_out` +Ruta al archivo csv de salida que contiene el DataFrame actualizado, por ejemplo, 'path/to/mutations_updated.csv'. Solo válido cuando se usa con `--update_df`. +Predeterminado: Ninguno -> el nuevo archivo csv se guardará en el mismo directorio que el DataFrame `mutations` con el apéndice '_updated' + +`-ts` `--translate_start` +(int o str) La posición en la secuencia de nucleótidos de entrada para comenzar a traducir, por ejemplo, 5. Si se proporciona una cadena, debe corresponder a un nombre de columna en `mutations` que contenga las posiciones de inicio del marco de lectura abierto para cada secuencia/mutación. Solo válido cuando se usa con `--translate`. +Predeterminado: traduce desde el principio de cada secuencia + +`-te` `--translate_end` +(int o str) La posición en la secuencia de nucleótidos de entrada para finalizar la traducción, por ejemplo, 35. Si se proporciona una cadena, debe corresponder a un nombre de columna en `mutations` que contenga las posiciones de fin del marco de lectura abierto para cada secuencia/mutación. Solo válido cuando se usa con `--translate`. +Predeterminado: traduce hasta el final de cada secuencia + +**Banderas opcionales para modificar salida adicional** +`-udf` `--update_df` +Actualiza el DataFrame de entrada `mutations` para incluir columnas adicionales con el tipo de mutación, la secuencia de nucleótidos de tipo salvaje y la secuencia de nucleótidos mutante (solo válido si `mutations` es un archivo .csv o .tsv). + +`-sfs` `--store_full_sequences` +Incluye las secuencias completas de tipo salvaje y mutantes en el DataFrame actualizado `mutations` (no solo la sub-secuencia con flancos de longitud k). Solo válido cuando se usa con `--update_df`. + +`-tr` `--translate` +Agrega columnas adicionales al DataFrame actualizado `mutations` que contienen las secuencias de aminoácidos de tipo salvaje y mutantes. Solo válido cuando se usa con `--store_full_sequences`. + +**Argumentos generales opcionales** `-o` `--out` Ruta al archivo FASTA de salida que contiene las secuencias mutadas, por ejemplo, 'path/to/output_fasta.fa'. -Por defecto: `None` -> devuelve una lista de las secuencias mutadas a la salida estándar. -Los identificadores (después del '>') de las secuencias mutadas en el FASTA de salida serán '>[seq_ID]_[mut_ID]'. +Predeterminado: Ninguno -> devuelve una lista de las secuencias mutadas a la salida estándar. +Los identificadores (que siguen al '>') de las secuencias mutadas en el FASTA de salida serán '>[seq_ID]_[mut_ID]'. -**Flags** +**Banderas generales opcionales** `-q` `--quiet` -Solo para Terminal. Impide la información de progreso de ser exhibida durante la ejecución del programa. -Para Python, usa `verbose=False` para imipidir la información de progreso de ser exhibida durante la ejecución del programa. +Solo en línea de comandos. Previene que se muestre información de progreso. +Python: Usa `verbose=False` para prevenir que se muestre información de progreso. + ### Ejemplos ```bash @@ -104,3 +159,89 @@ gget mutate ATCGCTAAGCT TAGCTA -m 'c.1_3inv' -k 3 gget.mutate(["ATCGCTAAGCT", "TAGCTA"], "c.1_3inv", k=3) ``` → Devuelve ['CTAGCT', 'GATCTA']. + +
+ +**Agregar mutaciones a un genoma completo con salida extendida** +Entrada principal: +- información de mutación como un CSV de `mutations` (teniendo `seq_id_column` que contenga información de cromosoma, y `mut_column` que contenga información de mutación con respecto a las coordenadas del genoma) +- el genoma como el archivo `sequences` + +Dado que estamos pasando la ruta a un archivo gtf al argumento `gtf`, se respetarán los límites de los transcritos (el genoma se dividirá en transcritos). `gtf_transcript_id_column` especifica el nombre de la columna en `mutations` que contiene los IDs de los transcritos correspondientes a los IDs de transcritos en el archivo `gtf`. + +El argumento `optimize_flanking_regions` maximiza la longitud de las secuencias resultantes que contienen la mutación manteniendo la especificidad (ningún k-mer de tipo salvaje se mantendrá). + +`update_df` activa la creación de un nuevo archivo CSV con información actualizada sobre cada secuencia de entrada y salida. Este nuevo archivo CSV se guardará como `update_df_out`. Dado que `store_full_sequences` está activado, este nuevo archivo CSV no solo contendrá las secuencias de salida (restringidas en tamaño por las regiones flanqueantes de tamaño `k`), sino también las secuencias completas de entrada y salida. Esto nos permite observar la mutación en el contexto de la secuencia completa. Por último, también estamos agregando las versiones traducidas de las secuencias completas mediante la activación de la bandera `translate`, para que podamos observar cómo cambia la secuencia de aminoácidos resultante. Los argumentos `translate_start` y `translate_end` especifican los nombres de las columnas en `mutations` que contienen las posiciones de inicio y fin del marco de lectura abierto (posiciones de inicio y fin para traducir la secuencia de nucleótidos a una secuencia de aminoácidos), respectivamente. + + +```bash +gget mutate \ + -m mutations_input.csv \ + -o mut_fasta.fa \ + -k 4 \ + -sic Chromosome \ + -mic Mutation \ + -gtf genome_annotation.gtf \ + -gtic Ensembl_Transcript_ID \ + -ofr \ + -update_df \ + -udf_o mutations_updated.csv \ + -sfs \ + -tr \ + -ts Translate_Start \ + -te Translate_End \ + genome_reference.fa +``` +```python +# Python +gget.mutate( + sequences="genome_reference.fa", + mutations="mutations_input.csv", + out="mut_fasta.fa", + k=4, + seq_id_column="Chromosome", + mut_column="Mutation", + gtf="genome_annotation.gtf", + gtf_transcript_id_column="Ensembl_Transcript_ID", + optimize_flanking_regions=True, + update_df=True, + update_df_out="mutations_updated.csv", + store_full_sequences=True, + translate=True, + translate_start="Translate_Start", + translate_end="Translate_End" +) +``` +→ Toma un genoma fasta ('genome_reference.fa') y un archivo gtf ('genome_annotation.gtf') (estos se pueden descargar usando [`gget ref`](ref.md)), así como un archivo 'mutations_input.csv' que contiene: +``` +| Chromosome | Mutation | Ensembl_Transcript_ID | Translate_Start | Translate_End | +|------------|-------------------|------------------------|-----------------|---------------| +| 1 | g.224411A>C | ENST00000193812 | 0 | 100 | +| 8 | g.25111del | ENST00000174411 | 0 | 294 | +| X | g.1011_1012insAA | ENST00000421914 | 9 | 1211 | +``` +→ Guarda el archivo 'mut_fasta.fa' que contiene: +``` +>1:g.224411A>C +TGCTCTGCT +>8:g.25111del +GAGTCGAT +>X:g.1011_1012insAA +TTAGAACTT +``` +→ Guarda el archivo 'mutations_updated.csv' que contiene: +``` + +| Chromosome | Mutation | Ensembl_Transcript_ID | mutation_type | wt_sequence | mutant_sequence | wt_sequence_full | mutant_sequence_full | wt_sequence_aa_full | mutant_sequence_aa_full | +|------------|-------------------|------------------------|---------------|-------------|-----------------|-------------------|----------------------|---------------------|-------------------------| +| 1 | g.224411A>C | ENSMUST00000193812 | Substitution | TGCTATGCT | TGCTCTGCT | ...TGCTATGCT... | ...TGCTCTGCT... | ...CYA... | ...CSA... | +| 8 | g.25111del | ENST00000174411 | Deletion | GAGTCCGAT | GAGTCGAT | ...GAGTCCGAT... | ...GAGTCGAT... | ...ESD... | ...ES... | +| X | g.1011_1012insAA | ENST00000421914 | Insertion | TTAGCTT | TTAGAACTT | ...TTAGCTT... | ...TTAGAACTT... | ...A... | ...EL... | + +``` + +# Citar +Si utiliza `gget mutate` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + diff --git a/docs/src/es/opentargets.md b/docs/src/es/opentargets.md new file mode 100644 index 00000000..638b8002 --- /dev/null +++ b/docs/src/es/opentargets.md @@ -0,0 +1,276 @@ +> Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. +# gget opentargets 🎯 +**Obtenga enfermedades o fármacos asociados con ciertos genes desde [OpenTargets](https://platform.opentargets.org/).** +Formato de salida: JSON/CSV (línea de comandos) o marco de datos (Python). + +Este módulo fue escrito por [Sam Wagenaar](https://github.com/techno-sam). + +**Argumento posicional** +`ens_id` +ID de gen Ensembl, por ejemplo, ENSG00000169194. + +**Argumentos opcionales** +`-r` `--resource` +Define el tipo de información a devolver en la salida. Predeterminado: 'diseases' (enfermedades). +Los recursos posibles son: + +| Recurso | Valor devuelto | Filtros válidos | Fuentes | +|--------------------|-------------------------------------------------------------------|---------------------------------------------------|----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------| +| `diseases` | Enfermedades asociadas | Ninguno | Varias:
- [Open Targets](https://genetics.opentargets.org/)
- [ChEMBL](https://www.ebi.ac.uk/chembl/)
- [Europe PMC](http://europepmc.org/)
`anatomical_system`
`organ` |
- [ExpressionAtlas](https://www.ebi.ac.uk/gxa/home)
- [HPA](https://www.proteinatlas.org/)
- [GTEx](https://www.gtexportal.org/home/)
`protein_b_id`
`gene_b_id` |
- [Open Targets](https://platform-docs.opentargets.org/target/molecular-interactions)
- [IntAct](https://platform-docs.opentargets.org/target/molecular-interactions#intact)
- [Signor](https://platform-docs.opentargets.org/target/molecular-interactions#signor)
- [Reactome](https://platform-docs.opentargets.org/target/molecular-interactions#reactome)
- [String](https://platform-docs.opentargets.org/target/molecular-interactions#string)
+ +**Obtener medicamentos asociados para un gen específico:** +```bash +gget opentargets ENSG00000169194 -r drugs -l 2 +``` +```python +# Python +import gget +gget.opentargets('ENSG00000169194', resource='drugs', limit=2) +``` + +→ Devuelve los 2 principales medicamentos asociados con el gen ENSG00000169194. + +| id | name | type | action_mechanism | description | synonyms | trade_names | disease_id | disease_name | trial_phase | trial_status | trial_ids | approved | +|---------------|--------------|----------|-------------------------------------|--------------------------------------------------------------|----------------------------------------------------|-----------------------|-------------|-------------------------------|-------------|--------------|---------------|----------| +| CHEMBL1743081 | TRALOKINUMAB | Antibody | Interleukin‑13 inhibitor | Antibody drug with a maximum clinical trial phase of IV ... | ['CAT-354', 'Tralokinumab'] | ['Adbry', 'Adtralza'] | EFO_0000274 | atopic eczema | 4 | | [] | True | +| CHEMBL4297864 | CENDAKIMAB | Antibody | Interleukin‑13 inhibitor | Antibody drug with a maximum clinical trial phase of III ... | [ABT-308, Abt-308, CC-93538, Cendakimab, RPC-4046] | [] | EFO_0004232 | eosinophilic esophagitis | 3 | Recruiting | [NCT04991935] | False | + +*Note: Los `trial_ids` devueltos son identificadores de [ClinicalTrials.gov](https://clinicaltrials.gov)* + +
+ +**Obtenga datos de trazabilidad para un gen específico:** +```bash +gget opentargets ENSG00000169194 -r tractability +``` +```python +# Python +import gget +gget.opentargets('ENSG00000169194', resource='tractability') +``` + +→ Devuelve datos de trazabilidad para el gen ENSG00000169194. + +| label | modality | +|-----------------------|----------------| +| High-Quality Pocket | Small molecule | +| Approved Drug | Antibody | +| GO CC high conf | Antibody | +| UniProt loc med conf | Antibody | +| UniProt SigP or TMHMM | Antibody | + +
+ +**Obtenga respuestas farmacogenéticas para un gen específico:** +```bash +gget opentargets ENSG00000169194 -r pharmacogenetics -l 1 +``` +```python +# Python +import gget +gget.opentargets('ENSG00000169194', resource='pharmacogenetics', limit=1) +``` + +→ Devuelve respuestas farmacogenéticas para el gen ENSG00000169194. + +| rs_id | genotype_id | genotype | variant_consequence_id | variant_consequence_label | drugs | phenotype | genotype_annotation | response_category | direct_target | evidence_level | source | literature | +|-----------|-------------------|----------|------------------------|---------------------------|-----------------------------------------------------------------------------------------------------------------------------------------|-------------------------------------------------------------------------|--------------------------------------------------------------------------------------------------------------|-------------------|---------------|----------------|----------|------------| +| rs1295686 | 5_132660151_T_T,T | TT | SO:0002073 | no_sequence_alteration | id name
0 None hepatitis vaccines | increased risk for non‑immune response to the hepatitis B vaccine | Patients with the TT genotype may be at increased risk for non-immune response to the hepatitis B vaccine... | efficacy | False | 3 | pharmgkb | [21111021] | + +*Note: Los identificadores de `literature` devueltos son identificadores de [PMC de Europa](https://europepmc.org/article/med/)* + +
+ +**Obtenga tejidos donde un gen se expresa más:** +```bash +gget opentargets ENSG00000169194 -r expression -l 2 +``` +```python +# Python +import gget +gget.opentargets('ENSG00000169194', resource='expression', limit=2) +``` + +→ Devuelve los 2 tejidos principales donde se expresa más el gen ENSG00000169194. + +| tissue_id | tissue_name | rna_zscore | rna_value | rna_unit | rna_level | anatomical_systems | organs | +|----------------|---------------------------------------|------------|-----------|----------|-----------|----------------------------------------------------------------------|--------------------------------------------------------| +| UBERON_0000473 | testis | 5 | 1026 | | 3 | [reproductive system] | [reproductive organ, reproductive structure] | +| CL_0000542 | EBV‑transformed lymphocyte | 1 | 54 | | 2 | [hemolymphoid system, immune system, lymphoid system] | [immune organ] | + +
+ +**Obtenga datos sobre el efecto de la enfermedad genética de DepMap para un gen específico:** +```bash +gget opentargets ENSG00000169194 -r depmap +``` +```python +# Python +import gget +gget.opentargets('ENSG00000169194', resource='depmap') +``` + +→ Devuelve datos del efecto de la enfermedad del gen DepMap para el gen ENSG00000169194. + +| depmap_id | expression | effect | tissue_id | tissue_name | cell_line_name | disease_cell_line_id | disease_name | mutation | +|------------------|------------|----------|----------------|-------------|----------------|----------------------|----------------------|----------| +| ACH‑001532 | 0.176323 | 0.054950 | UBERON_0002113 | kidney | JMU-RTK-2 | None | Rhabdoid Cancer | None | + +
+ +**Obtener interacciones proteína-proteína para un gen específico:** +```bash +gget opentargets ENSG00000169194 -r interactions -l 2 +``` +```python +# Python +import gget +gget.opentargets('ENSG00000169194', resource='interactions', limit=2) +``` + +→ Devuelve las 2 interacciones proteína-proteína principales para el gen ENSG00000169194. + +| evidence_score | evidence_count | source_db | protein_a_id | gene_a_id | gene_a_symbol | role_a | taxon_a | protein_b_id | gene_b_id | gene_b_symbol | role_b | taxon_b | +|----------------|----------------|-----------|-----------------|-----------------|---------------|-----------------------|---------|-----------------|-----------------|---------------|-----------------------|---------| +| 0.999 | 3 | string | ENSP00000304915 | ENSG00000169194 | IL13 | unspecified role | 9606 | ENSP00000379111 | ENSG00000077238 | IL4R | unspecified role | 9606 | +| 0.999 | 3 | string | ENSP00000304915 | ENSG00000169194 | IL13 | unspecified role | 9606 | ENSP00000360730 | ENSG00000131724 | IL13RA1 | unspecified role | 9606 | + +
+ +**Obtenga interacciones proteína-proteína para un gen específico, filtrando por ID de proteínas y genes:** +```bash +gget opentargets ENSG00000169194 -r interactions -fpa P35225 --filter_gene_b ENSG00000077238 +``` +```python +# Python +import gget +gget.opentargets('ENSG00000169194', resource='interactions', filters={'protein_a_id': 'P35225', 'gene_b_id': 'ENSG00000077238'}) +``` + +→ Devuelve interacciones proteína-proteína para el gen ENSG00000169194, donde la primera proteína es P35225 **y** el segundo gen es ENSG00000077238: + +| evidence_score | evidence_count | source_db | protein_a_id | gene_a_id | gene_a_symbol | role_a | taxon_a | protein_b_id | gene_b_id | gene_b_symbol | role_b | taxon_b | +|----------------|----------------|-----------|--------------|-----------------|---------------|-----------------------|---------|--------------|-----------------|---------------|-----------------------|---------| +| None | 3 | reactome | P35225 | ENSG00000169194 | IL13 | unspecified role | 9606 | P24394 | ENSG00000077238 | IL4R | unspecified role | 9606 | +| None | 2 | signor | P35225 | ENSG00000169194 | IL13 | regulator | 9606 | P24394 | ENSG00000077238 | IL4R | regulator target | 9606 | + +
+ +**Obtenga interacciones proteína-proteína para un gen específico, filtrando por ID de proteína o gen:** +```bash +gget opentargets ENSG00000169194 -r interactions -fpa P35225 --filter_gene_b ENSG00000077238 ENSG00000111537 --or -l 5 +``` +```python +# Python +import gget +gget.opentargets( + 'ENSG00000169194', + resource='interactions', + filters={'protein_a_id': 'P35225', 'gene_b_id': ['ENSG00000077238', 'ENSG00000111537']}, + filter_mode='or', + limit=5 +) +``` + +→ Devuelve interacciones proteína-proteína para el gen ENSG00000169194, donde la primera proteína es P35225 **o** el segundo gen es ENSG00000077238 o ENSG00000111537. +| evidence_score | evidence_count | source_db | protein_a_id | gene_a_id | gene_a_symbol | role_a | taxon_a | protein_b_id | gene_b_id | gene_b_symbol | role_b | taxon_b | +|----------------|----------------|-----------|-----------------|-----------------|---------------|-----------------------|---------|-----------------|-----------------|---------------|-----------------------|---------| +| 0.999 | 3 | string | ENSP00000304915 | ENSG00000169194 | IL13 | unspecified role | 9606 | ENSP00000379111 | ENSG00000077238 | IL4R | unspecified role | 9606 | +| 0.961 | 2 | string | ENSP00000304915 | ENSG00000169194 | IL13 | unspecified role | 9606 | ENSP00000229135 | ENSG00000111537 | IFNG | unspecified role | 9606 | +| 0.800 | 9 | intact | P35225 | ENSG00000169194 | IL13 | unspecified role | 9606 | Q14627 | ENSG00000123496 | IL13RA2 | unspecified role | 9606 | +| 0.740 | 6 | intact | P35225 | ENSG00000169194 | IL13 | unspecified role | 9606 | P78552 | ENSG00000131724 | IL13RA1 | unspecified role | 9606 | +| 0.400 | 1 | intact | P35225 | ENSG00000169194 | IL13 | unspecified role | 9606 | Q86XT9 | ENSG00000149932 | TMEM219 | stimulator | 9606 | + + + +#### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget opentargets` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Ochoa D, Hercules A, Carmona M, Suveges D, Baker J, Malangone C, Lopez I, Miranda A, Cruz-Castillo C, Fumis L, Bernal-Llinares M, Tsukanov K, Cornu H, Tsirigos K, Razuvayevskaya O, Buniello A, Schwartzentruber J, Karim M, Ariano B, Martinez Osorio RE, Ferrer J, Ge X, Machlitt-Northen S, Gonzalez-Uriarte A, Saha S, Tirunagari S, Mehta C, Roldán-Romero JM, Horswell S, Young S, Ghoussaini M, Hulcoop DG, Dunham I, McDonagh EM. The next-generation Open Targets Platform: reimagined, redesigned, rebuilt. Nucleic Acids Res. 2023 Jan 6;51(D1):D1353-D1359. doi: [10.1093/nar/gkac1046](https://doi.org/10.1093/nar/gkac1046). PMID: 36399499; PMCID: PMC9825572. diff --git a/docs/src/es/pdb.md b/docs/src/es/pdb.md index 0d36800f..c0205fc0 100644 --- a/docs/src/es/pdb.md +++ b/docs/src/es/pdb.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget pdb 🔮 +# gget pdb 🔮 Obtenga la estructura o los metadatos de una proteína usando data de [RCSB Protein Data Bank (PDB)](https://www.rcsb.org/). Regresa: El archivo 'pdb' se regresa en formato PDB. Todos los demás datos se regresan en formato JSON. @@ -77,3 +77,10 @@ gget.pdb("7CT5", save=True) → Este caso de uso ejemplifica cómo encontrar archivos PDB para un análisis comparativo de la estructura de las proteínas asociado con IDs de Ensembl o secuencias de aminoácidos. Los archivos PDB obtenidos también se pueden comparar con las estructuras predichas generadas por [`gget alphafold`](alphafold.md). Los archivos PDB se pueden ver de forma interactiva en 3D [aquí](https://rcsb.org/3d-view), o usando programas como [PyMOL](https://pymol.org/) o [Blender](https://www.blender.org/). Múltiple archivos PDB se pueden visualizar para comparación [aquí](https://rcsb.org/alignment). #### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget pdb` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000 Jan 1;28(1):235-42. doi: [10.1093/nar/28.1.235](https://doi.org/10.1093/nar/28.1.235). PMID: 10592235; PMCID: PMC102472. diff --git a/docs/src/es/quick_start_guide.md b/docs/src/es/quick_start_guide.md index 0753b190..a828ecf7 100644 --- a/docs/src/es/quick_start_guide.md +++ b/docs/src/es/quick_start_guide.md @@ -1,4 +1,4 @@ -## 🪄 Guía de inicio rápido +# 🪄 Guía de inicio rápido Terminal: ```bash # Obtenga todos los FTP de anotaciones y referencias de Homo sapiens de la última versión de Ensembl diff --git a/docs/src/es/ref.md b/docs/src/es/ref.md index cdd6a0d5..4ef5b7c9 100644 --- a/docs/src/es/ref.md +++ b/docs/src/es/ref.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget ref 📖 +# gget ref 📖 Obtenga enlaces FTP y sus respectivos metadatos (o use la bandera `ftp` para regresar solo los enlaces) para referenciar genomas y anotaciones de [Ensembl](https://www.ensembl.org/). Regresa: Resultados en formato JSON. @@ -23,6 +23,9 @@ Las entradas posibles son uno solo o una combinación de las siguientes (como li `-r` `--release` Define el número de versión de Ensembl desde el que se obtienen los archivos, p. ej. 104. Default: latest Ensembl release. +`-od` `--out_dir` +Ruta al directorio donde se guardarán los archivos FTP, p. ruta/al/directorio/. Por defecto: directorio de trabajo actual. + `-o` `--out` Ruta al archivo en el que se guardarán los resultados, p. ej. ruta/al/directorio/resultados.json. Por defecto: salida estándar (STDOUT). Para Python, usa `save=True` para guardar los resultados en el directorio de trabajo actual. @@ -94,3 +97,10 @@ gget.ref("homo_sapiens", which=["gtf", "dna"]) ``` #### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget ref` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. diff --git a/docs/src/es/search.md b/docs/src/es/search.md index 0899524d..ae6c0d57 100644 --- a/docs/src/es/search.md +++ b/docs/src/es/search.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget search 🔎 +# gget search 🔎 Obtenga genes y transcripciones de [Ensembl](https://www.ensembl.org/) usando términos de búsqueda de forma libre. Los resultados se comparan según las secciones "nombre del gen" y "descripción" en la base de datos de Ensembl. `gget` versión >= 0.27.9 también incluye resultados que coinciden con la sección "sinónimo" de Ensembl. Regresa: Resultados en formato JSON (Terminal) o Dataframe/CSV (Python). @@ -73,3 +73,10 @@ gget.search(["gaba", "gamma-aminobutyric"], "homo_sapiens") | . . . | . . . | . . . | . . . | . . . | . . . | #### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget search` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606. diff --git a/docs/src/es/seq.md b/docs/src/es/seq.md index c17eb865..d9c79cf2 100644 --- a/docs/src/es/seq.md +++ b/docs/src/es/seq.md @@ -49,3 +49,12 @@ gget.seq("ENSG00000034713", translate=True, isoforms=True) → Regresa las secuencias de aminoácidos de todas las transcripciones conocidas de ENSG00000034713 en formato FASTA. #### [Más ejemplos](https://github.com/pachterlab/gget_examples) + +# Citar +Si utiliza `gget seq` en una publicación, favor de citar los siguientes artículos: + +- Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. [https://doi.org/10.1093/bioinformatics/btac836](https://doi.org/10.1093/bioinformatics/btac836) + +- Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, Becker A, Bennett R, Berry A, Bhai J, Bhurji SK, Bignell A, Boddu S, Branco Lins PR, Brooks L, Ramaraju SB, Charkhchi M, Cockburn A, Da Rin Fiorretto L, Davidson C, Dodiya K, Donaldson S, El Houdaigui B, El Naboulsi T, Fatima R, Giron CG, Genez T, Ghattaoraya GS, Martinez JG, Guijarro C, Hardy M, Hollis Z, Hourlier T, Hunt T, Kay M, Kaykala V, Le T, Lemos D, Marques-Coelho D, Marugán JC, Merino GA, Mirabueno LP, Mushtaq A, Hossain SN, Ogeh DN, Sakthivel MP, Parker A, Perry M, Piližota I, Prosovetskaia I, Pérez-Silva JG, Salam AIA, Saraiva-Agostinho N, Schuilenburg H, Sheppard D, Sinha S, Sipos B, Stark W, Steed E, Sukumaran R, Sumathipala D, Suner MM, Surapaneni L, Sutinen K, Szpak M, Tricomi FF, Urbina-Gómez D, Veidenberg A, Walsh TA, Walts B, Wass E, Willhoft N, Allen J, Alvarez-Jarreta J, Chakiachvili M, Flint B, Giorgetti S, Haggerty L, Ilsley GR, Loveland JE, Moore B, Mudge JM, Tate J, Thybert D, Trevanion SJ, Winterbottom A, Frankish A, Hunt SE, Ruffier M, Cunningham F, Dyer S, Finn RD, Howe KL, Harrison PW, Yates AD, Flicek P. Ensembl 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D933-D941. doi: [10.1093/nar/gkac958](https://doi.org/10.1093/nar/gkac958). PMID: 36318249; PMCID: PMC9825606 + +- The UniProt Consortium , UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Research, Volume 51, Issue D1, 6 January 2023, Pages D523–D531, [https://doi.org/10.1093/nar/gkac1052](https://doi.org/10.1093/nar/gkac1052) diff --git a/docs/src/es/setup.md b/docs/src/es/setup.md index 8fce8889..ecd72bef 100644 --- a/docs/src/es/setup.md +++ b/docs/src/es/setup.md @@ -1,5 +1,5 @@ > Parámetros de Python són iguales a los parámetros largos (`--parámetro`) de Terminal, si no especificado de otra manera. Banderas son parámetros de verdadero o falso (True/False) en Python. El manuál para cualquier modulo de gget se puede llamar desde la Terminal con la bandera `-h` `--help`. -## gget setup 🔧 +# gget setup 🔧 Función para instalar/descargar dependencias de terceros para un módulo de gget. diff --git a/docs/src/es/updates.md b/docs/src/es/updates.md index a5c8d304..eb8cde3c 100644 --- a/docs/src/es/updates.md +++ b/docs/src/es/updates.md @@ -1,4 +1,23 @@ ## ✨ ¡Lo más reciente! +**Versión ≥ 0.29.0** (25 de septiembre de 2024): +- **Nuevos módulos:** + - [`gget cbio`](cbio.md) + - [`gget opentargets`](opentargets.md) + - [`gget bgee`](bgee.md) +- **[`gget enrichr`](./enrichr.md) ahora también soporta especies además de humano y ratón (mosca, levadura, gusano y pez) a través de [modEnrichR](https://maayanlab.cloud/modEnrichr/)** +- [`gget mutate`](./mutate.md): + `gget mutate` ahora fusionará secuencias idénticas en el archivo final por defecto. La creación de mutaciones fue vectorizada para disminuir el tiempo de ejecución. Se mejoró la verificación de la secuencia flanqueante para mutaciones no sustitutivas para asegurarse de que no se retenga ningún kmer silvestre en la secuencia que contiene la mutación. Se agregó varios nuevos argumentos para personalizar la generación de secuencias y la salida. +- [`gget cosmic`](./cosmic.md): + Se agregó soporte para pantallas de genes así como dirigidas. El archivo CSV creado para gget mutate ahora también contiene información sobre mutaciones de proteínas. +- [`gget ref`](./ref.md): + Se agregó opción de archivo de salida. +- [`gget info`](./info.md) y [`gget seq`](./seq.md): + Se cambió a la API POST de Ensembl para aumentar la velocidad (nada cambia en el front end). +- Otros cambios "detrás de escena": + - Pruebas unitarias reorganizadas para aumentar la velocidad y disminuir el código + - Requisitos actualizados para [permitir versiones más nuevas de mysql-connector](https://github.com/pachterlab/gget/pull/159) + - [Soporte para Numpy>= 2.0](https://github.com/pachterlab/gget/issues/157) + **Versión ≥ 0.28.6 (2 de junio de 2024):** - **Nuevo módulo: [`gget mutate`](./mutate.md)** - [`gget cosmic`](./cosmic.md): Ahora puedes descargar bases de datos completas de COSMIC utilizando el argumento `download_cosmic` diff --git a/gget/__init__.py b/gget/__init__.py index e0b42da6..74ceb95b 100644 --- a/gget/__init__.py +++ b/gget/__init__.py @@ -16,6 +16,7 @@ from .gget_diamond import diamond from .gget_cosmic import cosmic from .gget_mutate import mutate +from .gget_dataverse import dataverse from .gget_opentargets import opentargets from .gget_cbio import cbio_plot, cbio_search from .gget_bgee import bgee diff --git a/gget/constants.py b/gget/constants.py index a61f9a11..4eac22d6 100644 --- a/gget/constants.py +++ b/gget/constants.py @@ -63,6 +63,9 @@ COSMIC_GET_URL = "https://cancer.sanger.ac.uk/cosmic/search/" COSMIC_RELEASE_URL = "https://cancer.sanger.ac.uk/cosmic/release_notes" +# Harvard dataverse API server +DATAVERSE_GET_URL = "https://dataverse.harvard.edu/api/access/datafile/" + # OpenTargets API endpoint OPENTARGETS_GRAPHQL_API = "https://api.platform.opentargets.org/api/v4/graphql" diff --git a/gget/gget_bgee.py b/gget/gget_bgee.py index 4a9c9f8f..ce9b8915 100644 --- a/gget/gget_bgee.py +++ b/gget/gget_bgee.py @@ -161,7 +161,7 @@ def _bgee_expression(gene_id, json=False, verbose=True): # noinspection PyShadowingBuiltins def bgee( gene_id, - type, + type="orthologs", json=False, verbose=True, ): @@ -169,7 +169,7 @@ def bgee( Get orthologs/expression data for a gene from Bgee (https://www.bgee.org/). Args: - type type of data to retrieve (expression or orthologs) + type type of data to retrieve ('expression' or 'orthologs') gene_id Ensembl gene ID json return JSON instead of DataFrame verbose log progress @@ -181,4 +181,4 @@ def bgee( elif type == "orthologs": return _bgee_orthologs(gene_id, json=json, verbose=verbose) else: - raise ValueError(f"Unknown type: {type}") + raise ValueError(f"Argument type should be 'expression' or 'orthologs', not '{type}'") diff --git a/gget/gget_cbio.py b/gget/gget_cbio.py index b55872cf..e3f38922 100644 --- a/gget/gget_cbio.py +++ b/gget/gget_cbio.py @@ -20,9 +20,9 @@ if not hasattr(pd.DataFrame, "map"): - logger.info( - "Old pandas version detected. Patching DataFrame.map to DataFrame.applymap" - ) + # logger.warning( + # "Old pandas version detected. Patching DataFrame.map to DataFrame.applymap" + # ) pd.DataFrame.map = pd.DataFrame.applymap diff --git a/gget/gget_cosmic.py b/gget/gget_cosmic.py index 41a35770..0f60ee82 100644 --- a/gget/gget_cosmic.py +++ b/gget/gget_cosmic.py @@ -26,11 +26,13 @@ def is_valid_email(email): return re.match(email_pattern, email) is not None -def download_reference(download_link, tar_folder_path, file_path, verbose): - email = input("Please enter your COSMIC email: ") +def download_reference(download_link, tar_folder_path, file_path, verbose, email = None, password = None): + if not email: + email = input("Please enter your COSMIC email: ") if not is_valid_email(email): raise ValueError("The email address is not valid.") - password = getpass.getpass("Please enter your COSMIC password: ") + if not password: + password = getpass.getpass("Please enter your COSMIC password: ") # Concatenate the email and password with a colon input_string = f"{email}:{password}\n" @@ -81,7 +83,7 @@ def download_reference(download_link, tar_folder_path, file_path, verbose): def select_reference( - mutation_class, reference_dir, grch_version, cosmic_version, verbose + mutation_class, reference_dir, grch_version, cosmic_version, verbose, email = None, password = None ): # if mutation_class == "transcriptome": # download_link = f"https://cancer.sanger.ac.uk/api/mono/products/v1/downloads/scripted?path=grch{grch_version}/cosmic/v{cosmic_version}/Cosmic_Genes_Fasta_v{cosmic_version}_GRCh{grch_version}.tar&bucket=downloads" @@ -145,13 +147,16 @@ def select_reference( overwrite = True if os.path.exists(file_path): - proceed = ( - input( - "The requested COSMIC database already exists at the destination. Would you like to overwrite the existing files (y/n)? " + if not email and not password: + proceed = ( + input( + "The requested COSMIC database already exists at the destination. Would you like to overwrite the existing files (y/n)? " + ) + .strip() + .lower() ) - .strip() - .lower() - ) + else: + proceed = "yes" if proceed in ["yes", "y"]: overwrite = True else: @@ -176,15 +181,18 @@ def select_reference( # Download full databases else: - proceed = ( - input( - "Downloading complete databases from COSMIC requires an account (https://cancer.sanger.ac.uk/cosmic/register; free for academic use, license for commercial use).\nWould you like to proceed (y/n)? " + if email and password: + proceed = "yes" + else: + proceed = ( + input( + "Downloading complete databases from COSMIC requires an account (https://cancer.sanger.ac.uk/cosmic/register; free for academic use, license for commercial use).\nWould you like to proceed (y/n)? " + ) + .strip() + .lower() ) - .strip() - .lower() - ) if proceed in ["yes", "y"]: - download_reference(download_link, tar_folder_path, file_path, verbose) + download_reference(download_link, tar_folder_path, file_path, verbose, email = email, password = password) else: raise KeyboardInterrupt( f"Database download canceled. Learn more about COSMIC at https://cancer.sanger.ac.uk/cosmic/download/cosmic." @@ -214,6 +222,11 @@ def cosmic( gget_mutate=True, keep_genome_info=False, remove_duplicates=False, + seq_id_column="seq_ID", + mutation_column="mutation", + mut_id_column="mutation_id", + email=None, + password=None, out=None, verbose=True, ): @@ -258,6 +271,11 @@ def cosmic( - gget_mutate (True/False) Whether to create a modified version of the database for use with gget mutate. Default: True - keep_genome_info (True/False) Whether to keep genome information (e.g. location of mutation in the genome) in the modified database for use with gget mutate. Default: False - remove_duplicates (True/False) Whether to remove duplicate rows from the modified database for use with gget mutate. Default: False + - seq_id_column (str) Name of the seq_id column in the csv file created by gget_mutate. Default: "seq_ID" + - mutation_column (str) Name of the mutation column in the csv file created by gget_mutate. Default: "mutation" + - mut_id_column (str) Name of the mutation_id column in the csv file created by gget_mutate. Default: "mutation_id" + - email (str) Email for COSMIC login. Helpful for avoiding required input upon running gget COSMIC. Default: None + - password (str) Password for COSMIC login. Helpful for avoiding required input upon running gget COSMIC, but password will be stored in plain text in the script. Default: None General args: - out (str) Path to the file (or folder when downloading databases with the download_cosmic flag) the results will be saved in, e.g. 'path/to/results.json'. @@ -287,8 +305,8 @@ def cosmic( f"Parameter 'mutation_class' must be one of the following: {', '.join(mut_class_allowed)}.\n" ) - grch_allowed = [37, 38] - if grch_version not in grch_allowed: + grch_allowed = ['37', '38'] + if str(grch_version) not in grch_allowed: raise ValueError( f"Parameter 'grch_version' must be one of the following: {', '.join(grch_allowed)}.\n" ) @@ -308,7 +326,7 @@ def cosmic( ## Download requested database mutation_tsv_file, overwrite = select_reference( - mutation_class, out, grch_version, cosmic_version, verbose + mutation_class, out, grch_version, cosmic_version, verbose, email = email, password = password ) if gget_mutate and overwrite: @@ -524,7 +542,14 @@ def cosmic( df = df.drop_duplicates(subset=["seq_ID", "mutation"], keep="first") df = df.drop(columns=["non_na_count"]) - mutate_csv_out = mutation_tsv_file.replace(".tsv", "_gget_mutate.csv") + if isinstance(seq_id_column, str) and seq_id_column != "seq_ID": + df.rename(columns={"seq_ID": seq_id_column}, inplace=True) + if isinstance(mutation_column, str) and mutation_column and mutation_column != "mutation": + df.rename(columns={"mutation": mutation_column}, inplace=True) + if isinstance(mut_id_column, str) and mut_id_column != "mutation_id": + df.rename(columns={"mutation_id": mut_id_column}, inplace=True) + + mutate_csv_out = mutation_tsv_file.replace(".tsv", "_mutation_workflow.csv") df.to_csv(mutate_csv_out, index=False) if verbose: diff --git a/gget/gget_dataverse.py b/gget/gget_dataverse.py new file mode 100644 index 00000000..7d63f58e --- /dev/null +++ b/gget/gget_dataverse.py @@ -0,0 +1,89 @@ +import os +import requests +from tqdm import tqdm +import pandas as pd +import pandas as pd +from .utils import print_sys +from .constants import DATAVERSE_GET_URL + +def dataverse_downloader(url, path, file_name): + """dataverse download helper with progress bar + + Args: + url (str): the url of the dataset to download + path (str): the path to save the dataset locally + file_name (str): the name of the file to save locally + """ + save_path = os.path.join(path, file_name) + response = requests.get(url, stream=True) + total_size_in_bytes = int(response.headers.get("content-length", 0)) + block_size = 1024 + progress_bar = tqdm(total=total_size_in_bytes, unit="iB", unit_scale=True) + with open(save_path, "wb") as file: + for data in response.iter_content(block_size): + progress_bar.update(len(data)) + file.write(data) + progress_bar.close() + + +def download_wrapper(entry, path, return_type=None): + """wrapper for downloading a dataset given the name and path, for csv,pkl,tsv or similar files + + Args: + entry (dict): the entry of the dataset to download. Must include 'id', 'name', 'type' keys + path (str): the path to save the dataset locally + return_type (str, optional): the return type. Defaults to None. Can be "url", "filename", or ["url", "filename"] + + Returns: + str: the exact dataset query name + """ + url = DATAVERSE_GET_URL + str(entry['id']) + + if not os.path.exists(path): + os.mkdir(path) + + filename = f"{entry['name']}.{entry['type']}" + + if os.path.exists(os.path.join(path, filename)): + print_sys(f"Found local copy for {entry['id']} datafile as {filename} ...") + os.path.join(path, filename) + else: + print_sys(f"Downloading {entry['id']} datafile as {filename} ...") + dataverse_downloader(url, path, filename) + + if return_type == "url": + return url + elif return_type == "filename": + return filename + elif return_type == ["url", "filename"]: + return url, filename + + +def dataverse(df, path, sep=","): + """download datasets from dataverse for a given dataframe + Input dataframe must have 'name', 'id', 'type' columns. + - 'name' is the dataset name for single file + - 'id' is the unique identifier for the file + - 'type' is the file type (e.g. csv, tsv, pkl) + + Args: + df (pd.DataFrame or str): the dataframe or path to the csv/tsv file + path (str): the path to save the dataset locally + """ + if type(df) == str: + if os.path.exists(df): + df = pd.read_csv(df, sep=sep) + else: + raise FileNotFoundError(f"File {df} not found") + elif type(df) == pd.DataFrame: + pass + else: + raise ValueError("Input must be a pandas dataframe or a path to a csv / tsv file") + + print_sys(f"Searching for {len(df)} datafiles in dataverse ...") + + # run the download wrapper for each entry in the dataframe + for _, entry in df.iterrows(): + download_wrapper(entry, path) + + print_sys(f"Download completed, saved to `{path}`.") \ No newline at end of file diff --git a/gget/gget_diamond.py b/gget/gget_diamond.py index a7a1710d..e738ff1c 100644 --- a/gget/gget_diamond.py +++ b/gget/gget_diamond.py @@ -25,6 +25,7 @@ def diamond( query, reference, + translated=False, diamond_db=None, sensitivity="very-sensitive", threads=1, @@ -39,6 +40,9 @@ def diamond( Args: - query Sequences (str or list) or path to FASTA file containing sequences to be aligned against the reference. - reference Reference sequences (str or list) or path to FASTA file containing reference sequences. + Set translated=True if reference sequences are amino acid sequences and query sequences are nucleotide sequences. + - translated True/False whether to perform translated alignment of nucleotide sequences to amino acid reference sequences. + Default: False. - diamond_db Path to save DIAMOND database created from reference. Default: None -> Temporary db file will be deleted after alignment or saved in 'out' if 'out' is provided. - sensitivity Sensitivity of DIAMOND alignment. @@ -117,15 +121,22 @@ def diamond( reference_file_w = reference_file.replace("/", "\\") output_w = output.replace("/", "\\") + if translated: + if verbose: + logger.info(f"Aligning nucleotide query to amino acid reference (blastx mode).") + diamond_program = "blastx" + else: + diamond_program = "blastp" + if platform.system() == "Windows": command = f"{DIAMOND} version \ && {DIAMOND_w} makedb --quiet --in {reference_file_w} --db {diamond_db_w} --threads {threads} \ - && {DIAMOND_w} blastp --outfmt 6 qseqid sseqid pident qlen slen length mismatch gapopen qstart qend sstart send evalue bitscore \ + && {DIAMOND_w} {diamond_program} --outfmt 6 qseqid sseqid pident qlen slen length mismatch gapopen qstart qend sstart send evalue bitscore \ --quiet --query {input_file_w} --db {reference_file_w} --out {output_w} --{sensitivity} --threads {threads} --ignore-warnings" else: command = f"'{DIAMOND}' version \ && '{DIAMOND}' makedb --quiet --in '{reference_file}' --db '{diamond_db}' --threads {threads} \ - && '{DIAMOND}' blastp --outfmt 6 qseqid sseqid pident qlen slen length mismatch gapopen qstart qend sstart send evalue bitscore \ + && '{DIAMOND}' {diamond_program} --outfmt 6 qseqid sseqid pident qlen slen length mismatch gapopen qstart qend sstart send evalue bitscore \ --quiet --query '{input_file}' --db '{reference_file}' --out '{output}' --{sensitivity} --threads {threads} --ignore-warnings" # Run DIAMOND diff --git a/gget/gget_mutate.py b/gget/gget_mutate.py index 496b2609..920bd3ac 100644 --- a/gget/gget_mutate.py +++ b/gget/gget_mutate.py @@ -22,6 +22,12 @@ mutation_pattern = r"(?:c|g)\.([0-9_\-\+\*]+)([a-zA-Z>]+)" # more complex: r'c\.([0-9_\-\+\*\(\)\?]+)([a-zA-Z>\(\)0-9]+)' +def reverse_complement(seq): + if pd.isna(seq): # Check if the sequence is NaN + return np.nan + complement = str.maketrans("ATCGNatcgn.*", "TAGCNtagcn.*") + return seq.translate(complement)[::-1] + # Get complement complement = { "A": "T", @@ -118,74 +124,43 @@ def convert_chromosome_value_to_int_when_possible(val): return val -def merge_gtf_transcript_locations_into_cosmic_csv( - mutations, gtf_path, gtf_transcript_id_column -): - gtf_df = pd.read_csv( - gtf_path, - sep="\t", - comment="#", - header=None, - names=[ - "seqname", - "source", - "feature", - "start", - "end", - "score", - "strand", - "frame", - "attribute", - ], - ) +def merge_gtf_transcript_locations_into_cosmic_csv(mutations, gtf_path, gtf_transcript_id_column): + gtf_df = pd.read_csv(gtf_path, sep='\t', comment='#', header=None, names=[ + 'seqname', 'source', 'feature', 'start', 'end', 'score', 'strand', 'frame', 'attribute']) - if "strand" in mutations.columns: - mutations.rename(columns={"strand": "strand_original"}, inplace=True) + if 'strand' in mutations.columns: + mutations.rename(columns={'strand': 'strand_original'}, inplace=True) - gtf_df = gtf_df[gtf_df["feature"] == "transcript"] + gtf_df = gtf_df[gtf_df['feature'] == 'transcript'] - gtf_df["transcript_id"] = gtf_df["attribute"].str.extract('transcript_id "([^"]+)"') + gtf_df['transcript_id'] = gtf_df['attribute'].str.extract('transcript_id "([^"]+)"') - assert len(gtf_df["transcript_id"]) == len( - set(gtf_df["transcript_id"]) - ), "Duplicate transcript_id values found!" + assert len(gtf_df['transcript_id']) == len(set(gtf_df['transcript_id'])), "Duplicate transcript_id values found!" # Filter out rows where transcript_id is NaN - gtf_df = gtf_df.dropna(subset=["transcript_id"]) - - gtf_df = gtf_df[["transcript_id", "start", "end", "strand"]].rename( - columns={ - "transcript_id": gtf_transcript_id_column, - "start": "start_transcript_position", - "end": "end_transcript_position", - } - ) + gtf_df = gtf_df.dropna(subset=['transcript_id']) - merged_df = pd.merge(mutations, gtf_df, on=gtf_transcript_id_column, how="left") + gtf_df = gtf_df[['transcript_id', 'start', 'end', 'strand']].rename( + columns={'transcript_id': gtf_transcript_id_column, 'start': 'start_transcript_position', 'end': 'end_transcript_position'}) + + merged_df = pd.merge(mutations, gtf_df, on=gtf_transcript_id_column, how='left') # Fill NaN values - merged_df["start_transcript_position"] = merged_df[ - "start_transcript_position" - ].fillna(0) - merged_df["end_transcript_position"] = merged_df["end_transcript_position"].fillna( - 9999999 - ) - merged_df["strand"] = merged_df["strand"].fillna(".") + merged_df['start_transcript_position'] = merged_df['start_transcript_position'].fillna(0) + merged_df['end_transcript_position'] = merged_df['end_transcript_position'].fillna(9999999) + merged_df['strand'] = merged_df['strand'].fillna('.') return merged_df - def get_sequence_length(seq_id, seq_dict): return len(seq_dict.get(seq_id, "")) - def get_nucleotide_at_position(seq_id, pos, seq_dict): full_seq = seq_dict.get(seq_id, "") if pos < len(full_seq): return full_seq[pos] return None - def translate_sequence(sequence, start, end): amino_acid_sequence = "" for i in range(start, end, 3): @@ -201,17 +176,14 @@ def translate_sequence(sequence, start, end): # def remove_all_but_first_gt(line): # return line[:1] + line[1:].replace(">", "") - def remove_gt_after_semicolon(line): - parts = line.split(";") + parts = line.split(';') # Remove '>' from the beginning of each part except the first part - parts = [parts[0]] + [part.lstrip(">") for part in parts[1:]] - return ";".join(parts) + parts = [parts[0]] + [part.lstrip('>') for part in parts[1:]] + return ';'.join(parts) -def wt_fragment_and_mutant_fragment_share_kmer( - mutated_fragment: str, wildtype_fragment: str, k: int -) -> bool: +def wt_fragment_and_mutant_fragment_share_kmer(mutated_fragment: str, wildtype_fragment: str, k: int) -> bool: if len(mutated_fragment) <= k: if mutated_fragment in wildtype_fragment: return True @@ -219,7 +191,7 @@ def wt_fragment_and_mutant_fragment_share_kmer( return False else: for mutant_position in range(len(mutated_fragment) - k): - mutant_kmer = mutated_fragment[mutant_position : mutant_position + k] + mutant_kmer = mutated_fragment[mutant_position:mutant_position + k] if mutant_kmer in wildtype_fragment: # wt_position = wildtype_fragment.find(mutant_kmer) return True @@ -261,9 +233,9 @@ def add_mutation_type(mutations, mut_column): return mutations -def extract_sequence(row, seq_dict, seq_id_column="seq_ID"): +def extract_sequence(row, seq_dict, seq_id_column = "seq_ID"): if pd.isna(row["start_mutation_position"]) or pd.isna(row["end_mutation_position"]): - return None + return None seq = seq_dict[row[seq_id_column]][ int(row["start_mutation_position"]) : int(row["end_mutation_position"]) + 1 ] @@ -356,22 +328,10 @@ def mutate( mut_column: str = "mutation", seq_id_column: str = "seq_ID", mut_id_column: Optional[str] = None, - gtf: Optional[str] = None, - gtf_transcript_id_column: Optional[str] = None, - k: int = 30, - min_seq_len: Optional[int] = None, - optimize_flanking_regions: bool = False, - remove_seqs_with_wt_kmers: bool = False, - max_ambiguous: Optional[int] = None, - merge_identical: bool = True, - update_df: bool = False, - update_df_out: Optional[str] = None, - store_full_sequences: bool = False, - translate: bool = False, - translate_start: Union[int, str, None] = None, - translate_end: Union[int, str, None] = None, + k: Optional[int] = None, out: Optional[str] = None, verbose: bool = True, + **kwargs, ): """ Takes in nucleotide sequences and mutations (in standard mutation annotation - see below) @@ -393,7 +353,7 @@ def mutate( NOTE: Only the letters until the first space or dot will be used as sequence identifiers - Version numbers of Ensembl IDs will be ignored. NOTE: When 'sequences' input is a genome, also see 'gtf' argument below. - + - mutations Path to csv or tsv file (str) (e.g., 'mutations.csv') or data frame (DataFrame object) containing information about the mutations in the following format: @@ -411,48 +371,49 @@ def mutate( Alternatively: Input mutation(s) as a string or list, e.g., 'c.2C>T' or ['c.2C>T', 'c.1A>C']. If a list is provided, the number of mutations must equal the number of input sequences. - + For more information on the standard mutation annotation, see https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1867422/. - + Additional input arguments: - mut_column (str) Name of the column containing the mutations to be performed in 'mutations'. Default: 'mutation'. - seq_id_column (str) Name of the column containing the IDs of the sequences to be mutated in 'mutations'. Default: 'seq_ID'. - mut_id_column (str) Name of the column containing the IDs of each mutation in 'mutations'. Default: Will use mut_column. - - gtf (str) Path to .gtf file. When providing a genome fasta file as input for 'sequences', you can provide a .gtf file here + - gtf (str) Path to .gtf file. When providing a genome fasta file as input for 'sequences', you can provide a .gtf file here and the input sequences will be defined according to the transcript boundaries. Default: None - gtf_transcript_id_column (str) Column name in the input 'mutations' file containing the transcript ID. In this case, column seq_id_column should contain the chromosome number. Required when 'gtf' is provided. Default: None Mutant sequence generation/filtering options: - - k (int) Length of sequences flanking the mutation. Default: 30. + - k (int) Length of sequences flanking the mutation. Default: None (take entire sequence). If k > total length of the sequence, the entire sequence will be kept. - - min_seq_len (int) Minimum length of the mutant output sequence. Mutant sequences smaller than this will be dropped. + - min_seq_len (int) Minimum length of the mutant output sequence. Mutant sequences smaller than this will be dropped. Default: None - - optimize_flanking_regions (True/False) Whether to remove nucleotides from either end of the mutant sequence to ensure (when possible) + - optimize_flanking_regions (True/False) Whether to remove nucleotides from either end of the mutant sequence to ensure (when possible) that the mutant sequence does not contain any k-mers also found in the wildtype/input sequence. Default: False - - remove_seqs_with_wt_kmers (True/False) Removes output sequences where at least one (k+1)-mer is also present in the wildtype/input sequence in the same region. + - remove_seqs_with_wt_kmers (True/False) Removes output sequences where at least one (k+1)-mer is also present in the wildtype/input sequence in the same region. If optimize_flanking_regions=True, only sequences for which a wildtpye kmer is still present after optimization will be removed. Default: False - max_ambiguous (int) Maximum number of 'N' characters allowed in the output sequence. Default: None (no 'N' filter will be applied) - - merge_identical (True/False) Whether to merge identical mutant sequences in the output (identical sequences will be merged by concatenating the sequence + - merge_identical (True/False) Whether to merge identical mutant sequences in the output (identical sequences will be merged by concatenating the sequence headers for all identical sequences). Default: True + - merge_identical_rc (True/False) Whether to merge identical sequences and their reverse complements in the output. Only effective when merge_identical is also True. Default: True # Optional arguments to generate additional output stored in a copy of the 'mutations' DataFrame - - update_df (True/False) Whether to update the input 'mutations' DataFrame to include additional columns with the mutation type, + - update_df (True/False) Whether to update the input 'mutations' DataFrame to include additional columns with the mutation type, wildtype nucleotide sequence, and mutant nucleotide sequence (only valid if 'mutations' is a csv or tsv file). Default: False - update_df_out (str) Path to output csv file containing the updated DataFrame. Only valid if update_df=True. Default: None -> the new DataFrame will be saved in the same directory as the 'mutations' DataFrame with appendix '_updated' - - store_full_sequences (True/False) Whether to also include the complete wildtype and mutant sequences in the updated 'mutations' DataFrame (not just the sub-sequence with + - store_full_sequences (True/False) Whether to also include the complete wildtype and mutant sequences in the updated 'mutations' DataFrame (not just the sub-sequence with k-length flanks). Only valid if update_df=True. Default: False - - translate (True/False) Add additional columns to the 'mutations' DataFrame containing the wildtype and mutant amino acid sequences. + - translate (True/False) Add additional columns to the 'mutations' DataFrame containing the wildtype and mutant amino acid sequences. Only valid if store_full_sequences=True. Default: False - - translate_start (int | str | None) The position in the input nucleotide sequence to start translating. If a string is provided, it should correspond + - translate_start (int | str | None) The position in the input nucleotide sequence to start translating. If a string is provided, it should correspond to a column name in 'mutations' containing the open reading frame start positions for each sequence/mutation. Only valid if translate=True. Default: None (translate from the beginning of the sequence) - - translate_end (int | str | None) The position in the input nucleotide sequence to end translating. If a string is provided, it should correspond + - translate_end (int | str | None) The position in the input nucleotide sequence to end translating. If a string is provided, it should correspond to a column name in 'mutations' containing the open reading frame end positions for each sequence/mutation. Only valid if translate=True. Default: None (translate from to the end of the sequence) - + # General arguments: - out (str) Path to output fasta file containing the mutated sequences, e.g., 'path/to/output_fasta.fa'. Default: None -> returns a list of the mutated sequences to standard out. @@ -462,16 +423,21 @@ def mutate( Saves mutated sequences in fasta format (or returns a list containing the mutated sequences if out=None). """ + if kwargs.get("gtf") or kwargs.get("gtf_transcript_id_column") or kwargs.get("optimize_flanking_regions") or kwargs.get("remove_seqs_with_wt_kmers") or kwargs.get("min_seq_len") or kwargs.get("max_ambiguous") or kwargs.get("merge_identical") or kwargs.get("merge_identical_rc") or kwargs.get("update_df") or kwargs.get("update_df_out") or kwargs.get("store_full_sequences") or kwargs.get("translate") or kwargs.get("translate_start") or kwargs.get("translate_end"): + # print a log message and raise exception + logger.critical( + """ + It appears that you are passing in arguments that are not supported anymore in gget mutate. For use of these arguments, please check out https://github.com/pachterlab/kvar. + """ + ) + raise NotImplementedError + global intronic_mutations, posttranslational_region_mutations, unknown_mutations, uncertain_mutations, ambiguous_position_mutations, cosmic_incorrect_wt_base, mut_idx_outside_seq - columns_to_keep = [ - "header", - seq_id_column, - mut_column, - "mutation_type", - "wt_sequence", - "mutant_sequence", - ] + columns_to_keep = ["header", seq_id_column, mut_column, "mutation_type", "wt_sequence", "mutant_sequence"] + + if k is None: + k = 999999999 # take entire sequence by default # Load input sequences and their identifiers from fasta file if "." in sequences: @@ -506,18 +472,14 @@ def mutate( mutations = pd.read_csv(mutations) for col in mutations.columns: if col not in columns_to_keep: - columns_to_keep.append( - col - ) # append "mutation_aa", "gene_name", "mutation_id" + columns_to_keep.append(col) # append "mutation_aa", "gene_name", "mutation_id" elif isinstance(mutations, str) and mutations.endswith(".tsv"): mutations_path = mutations mutations = pd.read_csv(mutations, sep="\t") for col in mutations.columns: if col not in columns_to_keep: - columns_to_keep.append( - col - ) # append "mutation_aa", "gene_name", "mutation_id" + columns_to_keep.append(col) # append "mutation_aa", "gene_name", "mutation_id" # Handle mutations passed as a list elif isinstance(mutations, list): @@ -598,16 +560,10 @@ def mutate( mutations = mutations.dropna(subset=[seq_id_column]) # ensure seq_ID column is string type, and chromosome numbers don't have decimals - mutations[seq_id_column] = mutations[seq_id_column].apply( - convert_chromosome_value_to_int_when_possible - ) + mutations[seq_id_column] = mutations[seq_id_column].apply(convert_chromosome_value_to_int_when_possible) mutations = add_mutation_type(mutations, mut_column) - # Link sequences to their mutations using the sequence identifiers - if store_full_sequences: - mutations["wt_sequence_full"] = mutations[seq_id_column].map(seq_dict) - # Handle sequences that were not found based on their sequence IDs seqs_not_found = mutations[~mutations[seq_id_column].isin(seq_dict.keys())] if 0 < len(seqs_not_found) < 20: @@ -638,14 +594,16 @@ def mutate( ) total_mutations = mutations.shape[0] - - if mut_id_column is None: - mut_id_column = mut_column - mutations["mutant_sequence"] = "" - mutations["header"] = ( - ">" + mutations[seq_id_column] + ":" + mutations[mut_id_column] - ) + + if mut_id_column is not None: + mutations["header"] = ( + ">" + mutations[mut_id_column] + ) + else: + mutations["header"] = ( + ">" + mutations[seq_id_column] + ":" + mutations[mut_column] + ) # Calculate number of bad mutations uncertain_mutations = mutations[mut_column].str.contains(r"\?").sum() @@ -698,9 +656,7 @@ def mutate( mutations["end_mutation_position"] -= 1 # don't forget to increment by 1 later # Calculate sequence length - mutations["sequence_length"] = mutations[seq_id_column].apply( - lambda x: get_sequence_length(x, seq_dict) - ) + mutations["sequence_length"] = mutations[seq_id_column].apply(lambda x: get_sequence_length(x, seq_dict)) # Filter out mutations with positions outside the sequence index_error_mask = ( @@ -724,31 +680,6 @@ def mutate( duplication_mask = mutations["mutation_type"] == "duplication" inversion_mask = mutations["mutation_type"] == "inversion" - if remove_seqs_with_wt_kmers: - long_duplications = ( - (duplication_mask) - & ( - ( - mutations["end_mutation_position"] - - mutations["start_mutation_position"] - ) - >= k - ) - ).sum() - logger.info(f"Removing {long_duplications} duplications > k") - mutations = mutations[ - ~( - (duplication_mask) - & ( - ( - mutations["end_mutation_position"] - - mutations["start_mutation_position"] - ) - >= k - ) - ) - ] - # Create a mask for all non-substitution mutations non_substitution_mask = ( deletion_mask | delins_mask | insertion_mask | duplication_mask | inversion_mask @@ -756,16 +687,12 @@ def mutate( # Extract the WT nucleotides for the substitution rows from reference fasta (i.e., Ensembl) start_positions = mutations.loc[substitution_mask, "start_mutation_position"].values - + # Get the nucleotides at the start positions - wt_nucleotides_substitution = np.array( - [ - get_nucleotide_at_position(seq_id, pos, seq_dict) - for seq_id, pos in zip( - mutations.loc[substitution_mask, seq_id_column], start_positions - ) - ] - ) + wt_nucleotides_substitution = np.array([ + get_nucleotide_at_position(seq_id, pos, seq_dict) + for seq_id, pos in zip(mutations.loc[substitution_mask, seq_id_column], start_positions) + ]) mutations.loc[substitution_mask, "wt_nucleotides_ensembl"] = ( wt_nucleotides_substitution @@ -778,9 +705,8 @@ def mutate( ].str[0] congruent_wt_bases_mask = ( - mutations["wt_nucleotides_cosmic"] == mutations["wt_nucleotides_ensembl"] - ) | mutations[["wt_nucleotides_cosmic", "wt_nucleotides_ensembl"]].isna().any( - axis=1 + (mutations["wt_nucleotides_cosmic"] == mutations["wt_nucleotides_ensembl"]) | + mutations[["wt_nucleotides_cosmic", "wt_nucleotides_ensembl"]].isna().any(axis=1) ) cosmic_incorrect_wt_base = (~congruent_wt_bases_mask).sum() @@ -849,62 +775,7 @@ def mutate( axis=1 ) # don't forget to increment by 1 later on - if gtf is not None: - assert mutations_path.endswith(".csv") or mutations_path.endswith( - ".tsv" - ), "Mutations must be a CSV or TSV file" - if ( - "start_transcript_position" not in mutations.columns - and "end_transcript_position" not in mutations.columns - ): # * currently hard-coded column names, but optionally can be changed to arguments later - mutations = merge_gtf_transcript_locations_into_cosmic_csv( - mutations, gtf, gtf_transcript_id_column=gtf_transcript_id_column - ) - columns_to_keep.extend( - ["start_transcript_position", "end_transcript_position", "strand"] - ) - else: - logger.warning( - "Transcript positions already present in the input mutations file. Skipping GTF file merging." - ) - - # adjust start_transcript_position to be 0-index - mutations["start_transcript_position"] -= 1 - - mutations["start_kmer_position"] = mutations[ - ["start_kmer_position", "start_transcript_position"] - ].max(axis=1) - mutations["end_kmer_position"] = mutations[ - ["end_kmer_position", "end_transcript_position"] - ].min(axis=1) - - mut_apply = ( - (lambda *args, **kwargs: mutations.progress_apply(*args, **kwargs)) - if verbose - else mutations.apply - ) - - if update_df and store_full_sequences: - # Extract flank sequences - if verbose: - tqdm.pandas(desc="Extracting full left flank sequences") - - mutations["left_flank_region_full"] = mut_apply( - lambda row: seq_dict[row[seq_id_column]][ - 0 : row["start_mutation_position"] - ], - axis=1, - ) # ? vectorize - - if verbose: - tqdm.pandas(desc="Extracting full right flank sequences") - - mutations["right_flank_region_full"] = mut_apply( - lambda row: seq_dict[row[seq_id_column]][ - row["end_mutation_position"] + 1 : row["sequence_length"] - ], - axis=1, - ) # ? vectorize + mut_apply = (lambda *args, **kwargs: mutations.progress_apply(*args, **kwargs)) if verbose else mutations.apply if verbose: tqdm.pandas(desc="Extracting k-mer left flank sequences") @@ -941,52 +812,8 @@ def mutate( # To what extend the beginning of i overlaps with the beginning of d --> shave up to that many nucleotides off the beginning of r1 until k - len(r1) ≥ extent of overlap # To what extend the end of i overlaps with the beginning of d --> shave up to that many nucleotides off the end of r2 until k - len(r2) ≥ extent of overlap - if optimize_flanking_regions: - # Apply the function for beginning of mut_nucleotides with right_flank_region - mutations.loc[ - non_substitution_mask, "beginning_mutation_overlap_with_right_flank" - ] = mutations.loc[non_substitution_mask].apply( - calculate_beginning_mutation_overlap_with_right_flank, axis=1 - ) - - # Apply the function for end of mut_nucleotides with left_flank_region - mutations.loc[non_substitution_mask, "end_mutation_overlap_with_left_flank"] = ( - mutations.loc[non_substitution_mask].apply( - calculate_end_mutation_overlap_with_left_flank, axis=1 - ) - ) - - # Calculate k-len(flank) (see above instructions) - mutations.loc[non_substitution_mask, "k_minus_left_flank_length"] = ( - k - mutations.loc[non_substitution_mask, "left_flank_region"].apply(len) - ) - mutations.loc[non_substitution_mask, "k_minus_right_flank_length"] = ( - k - mutations.loc[non_substitution_mask, "right_flank_region"].apply(len) - ) - - mutations.loc[non_substitution_mask, "updated_left_flank_start"] = np.maximum( - mutations.loc[ - non_substitution_mask, "beginning_mutation_overlap_with_right_flank" - ] - - mutations.loc[non_substitution_mask, "k_minus_left_flank_length"], - 0, - ) - mutations.loc[non_substitution_mask, "updated_right_flank_end"] = np.maximum( - mutations.loc[non_substitution_mask, "end_mutation_overlap_with_left_flank"] - - mutations.loc[non_substitution_mask, "k_minus_right_flank_length"], - 0, - ) - - mutations["updated_left_flank_start"] = ( - mutations["updated_left_flank_start"].fillna(0).astype(int) - ) - mutations["updated_right_flank_end"] = ( - mutations["updated_right_flank_end"].fillna(0).astype(int) - ) - - else: - mutations["updated_left_flank_start"] = 0 - mutations["updated_right_flank_end"] = 0 + mutations["updated_left_flank_start"] = 0 + mutations["updated_right_flank_end"] = 0 # Create WT substitution k-mer sequences mutations.loc[substitution_mask, "wt_sequence"] = ( @@ -1001,9 +828,7 @@ def mutate( ].apply( lambda row: row["left_flank_region"][row["updated_left_flank_start"] :] + row["wt_nucleotides_ensembl"] - + row["right_flank_region"][ - : len(row["right_flank_region"]) - row["updated_right_flank_end"] - ], + + row["right_flank_region"][: len(row['right_flank_region']) - row["updated_right_flank_end"]], axis=1, ) @@ -1020,43 +845,10 @@ def mutate( ].apply( lambda row: row["left_flank_region"][row["updated_left_flank_start"] :] + row["mut_nucleotides"] - + row["right_flank_region"][ - : len(row["right_flank_region"]) - row["updated_right_flank_end"] - ], + + row["right_flank_region"][: len(row['right_flank_region']) - row["updated_right_flank_end"]], axis=1, ) - if remove_seqs_with_wt_kmers: - if verbose: - tqdm.pandas( - desc="Removing mutant fragments that share a kmer with wt fragments" - ) - - mutations["wt_fragment_and_mutant_fragment_share_kmer"] = mut_apply( - lambda row: wt_fragment_and_mutant_fragment_share_kmer( - mutated_fragment=row["mutant_sequence"], - wildtype_fragment=row["wt_sequence"], - k=k + 1, - ), - axis=1, - ) - - mutations_overlapping_with_wt = mutations[ - "wt_fragment_and_mutant_fragment_share_kmer" - ].sum() - - mutations = mutations[~mutations["wt_fragment_and_mutant_fragment_share_kmer"]] - - if update_df and store_full_sequences: - columns_to_keep.extend(["wt_sequence_full", "mutant_sequence_full"]) - - # Create full sequences (substitution and non-substitution) - mutations["mutant_sequence_full"] = ( - mutations["left_flank_region_full"] - + mutations["mut_nucleotides"] - + mutations["right_flank_region_full"] - ) - # Calculate k-mer lengths and report the distribution mutations["mutant_sequence_kmer_length"] = mutations["mutant_sequence"].apply( lambda x: len(x) if pd.notna(x) else 0 @@ -1064,49 +856,6 @@ def mutate( max_length = mutations["mutant_sequence_kmer_length"].max() - if min_seq_len: - rows_less_than_minimum = ( - mutations["mutant_sequence_kmer_length"] < min_seq_len - ).sum() - - mutations = mutations[mutations["mutant_sequence_kmer_length"] >= min_seq_len] - - if verbose: - logger.info( - f"Removed {rows_less_than_minimum} mutant kmers with length less than {min_seq_len}..." - ) - - if max_ambiguous is not None: - # Get number of 'N' or 'n' occuring in the sequence - mutations["num_N"] = mutations["mutant_sequence"].str.lower().str.count("n") - num_rows_with_N = (mutations["num_N"] > max_ambiguous).sum() - mutations = mutations[mutations["num_N"] <= max_ambiguous] - - if verbose: - logger.info( - f"Removed {num_rows_with_N} mutant kmers containing more than {max_ambiguous} 'N's..." - ) - - # Drop the 'num_N' column after filtering - mutations = mutations.drop(columns=["num_N"]) - - try: - # Create bins of width 5 from 0 to max_length - bins = range(0, max_length + 6, 5) - - # Bin the lengths and count the number of elements in each bin - binned_lengths = pd.cut( - mutations["mutant_sequence_kmer_length"], bins=bins, right=False - ) - bin_counts = binned_lengths.value_counts().sort_index() - - # Display the report - if verbose: - logger.debug("Report of the number of elements in each bin of width 5:") - logger.debug(bin_counts) - except Exception as e: - pass - # split_cols = mutations[mut_id_column].str.split("_", n=1, expand=True) # if split_cols.shape[1] == 1: @@ -1123,7 +872,7 @@ def mutate( # if remove_seqs_with_wt_kmers: # good_mutations = good_mutations - long_duplications - mutations_overlapping_with_wt - + # if min_seq_len: # good_mutations = good_mutations - rows_less_than_minimum @@ -1141,158 +890,15 @@ def mutate( {mut_idx_outside_seq} mutations with indices outside of the sequence length found ({mut_idx_outside_seq/total_mutations*100:.2f}%) """ - if remove_seqs_with_wt_kmers: - report += f"""{long_duplications} duplications longer than k found ({long_duplications/total_mutations*100:.2f}%) - {mutations_overlapping_with_wt} mutations with overlapping kmers found ({mutations_overlapping_with_wt/total_mutations*100:.2f}%) - """ - - if min_seq_len: - report += f"""{rows_less_than_minimum} mutations with fragment length < k found ({rows_less_than_minimum/total_mutations*100:.2f}%) - """ - - if max_ambiguous is not None: - report += f"""{num_rows_with_N} mutations with Ns found ({num_rows_with_N/total_mutations*100:.2f}%) - """ - if good_mutations != total_mutations: logger.warning(report) else: logger.info("All mutations correctly recorded") - if translate and update_df and store_full_sequences: - columns_to_keep.extend(["wt_sequence_aa_full", "mutant_sequence_aa_full"]) - - if not mutations_path: - assert ( - type(translate_start) != str and type(translate_end) != str - ), "translate_start and translate_end must be integers when translating sequences (or default None)." - if translate_start is None: - translate_start = 0 - if translate_end is None: - translate_end = mutations["sequence_length"][0] - - # combined_df['ORF'] = combined_df[translate_start] % 3 - - if verbose: - tqdm.pandas(desc="Translating WT amino acid sequences") - mutations["wt_sequence_aa_full"] = mutations[ - "wt_sequence_full" - ].progress_apply( - lambda x: translate_sequence( - x, start=translate_start, end=translate_end - ) - ) - else: - mutations["wt_sequence_aa_full"] = mutations["wt_sequence_full"].apply( - lambda x: translate_sequence( - x, start=translate_start, end=translate_end - ) - ) - - if verbose: - tqdm.pandas(desc="Translating mutant amino acid sequences") - - mutations["mutant_sequence_aa_full"] = mutations[ - "mutant_sequence_full" - ].progress_apply( - lambda x: translate_sequence( - x, start=translate_start, end=translate_end - ) - ) - - else: - mutations["mutant_sequence_aa_full"] = mutations[ - "mutant_sequence_full" - ].apply( - lambda x: translate_sequence( - x, start=translate_start, end=translate_end - ) - ) - - print(f"Translated mutated sequences: {mutations['wt_sequence_aa_full']}") - else: - if not translate_start: - translate_start = "translate_start" - - if not translate_end: - translate_end = "translate_end" - - if translate_start not in mutations.columns: - mutations["translate_start"] = 0 - - if translate_end not in mutations.columns: - mutations["translate_end"] = mutations["sequence_length"] - - if verbose: - tqdm.pandas(desc="Translating WT amino acid sequences") - - mutations["wt_sequence_aa_full"] = mut_apply( - lambda row: translate_sequence( - row["wt_sequence_full"], row[translate_start], row[translate_end] - ), - axis=1, - ) - - if verbose: - tqdm.pandas(desc="Translating mutant amino acid sequences") - - mutations["mutant_sequence_aa_full"] = mut_apply( - lambda row: translate_sequence( - row["mutant_sequence_full"], - row[translate_start], - row[translate_end], - ), - axis=1, - ) - mutations = mutations[columns_to_keep] - if merge_identical: - logger.info("Merging identical mutated sequences") - if update_df: - logger.warning( - "Merging identical mutated sequences can take a while if update_df=True since it will concatenate all MCRSs too)" - ) - mutations = ( - mutations.groupby("mutant_sequence", sort=False) - .agg( - lambda x: ";".join(x.astype(str)) - ) # Concatenate values with semicolons - .reset_index() - ) - - else: - mutations = ( - mutations.groupby("mutant_sequence", sort=False, group_keys=False)[ - "header" - ] - .apply(";".join) - .reset_index() - ) - - # apply remove_gt_after_semicolon to mutant_sequence - mutations["header"] = mutations["header"].apply(remove_gt_after_semicolon) - - # Calculate the number of semicolons in each entry - mutations["semicolon_count"] = mutations["header"].str.count(";") - - mutations["semicolon_count"] += 1 - - # Convert all 1 values to NaN - mutations["semicolon_count"] = mutations["semicolon_count"].replace(1, np.nan) - - # Take the sum across all rows of the new column - total_semicolons = int(mutations["semicolon_count"].sum()) - - mutations = mutations.drop(columns=["semicolon_count"]) - - if verbose: - logger.info( - f"{total_semicolons} identical mutated sequences were merged (headers were combined and separated using a semicolon (;). Occurences of identical mutated sequences may be reduced by increasing k." - ) - empty_kmer_count = (mutations["mutant_sequence"] == "").sum() - + if empty_kmer_count > 0 and verbose: logger.warning( f"{empty_kmer_count} mutated sequences were empty and were not included in the output." @@ -1300,26 +906,7 @@ def mutate( mutations = mutations[mutations["mutant_sequence"] != ""] - mutations["header"] = mutations["header"].str[1:] # remove the > character - - if update_df: - logger.info("Saving dataframe with updated mutation info...") - saved_updated_df = True - logger.warning( - "File size can be very large if the number of mutations is large." - ) - if not update_df_out: - if not mutations_path: - logger.warning( - "mutations_path must be provided if update_df is True and update_df_out is not provided." - ) - saved_updated_df = False - else: - base_name, ext = os.path.splitext(mutations_path) - update_df_out = f"{base_name}_updated{ext}" - if saved_updated_df: - mutations.to_csv(update_df_out, index=False) - print(f"Updated mutation info has been saved to {update_df_out}") + mutations['header'] = mutations['header'].str[1:] # remove the > character mutations["fasta_format"] = ( ">" + mutations["header"] + "\n" + mutations["mutant_sequence"] + "\n" diff --git a/gget/main.py b/gget/main.py index 72a7966e..754555d6 100644 --- a/gget/main.py +++ b/gget/main.py @@ -36,6 +36,7 @@ from .gget_diamond import diamond from .gget_cosmic import cosmic from .gget_mutate import mutate +from .gget_dataverse import dataverse from .gget_opentargets import opentargets, OPENTARGETS_RESOURCES from .gget_cbio import cbio_plot, cbio_search from .gget_bgee import bgee @@ -69,6 +70,7 @@ def int_or_str(value): return int(value) except ValueError: return value +from .gget_dataverse import dataverse def main(): @@ -456,7 +458,15 @@ def main(): type=str, nargs="+", required=True, - help="Reference sequences (str or list) or path to FASTA file containing reference sequences.", + help="Reference sequences (str or list) or path to FASTA file containing reference sequences. Add `-x` flag if reference sequences are amino acid sequences and query sequences are nucleotide sequences.", + ) + parser_diamond.add_argument( + "-x", + "--translated", + default=False, + action="store_true", + required=False, + help="Perform translated alignment of nucleotide sequences to amino acid reference sequences.", ) parser_diamond.add_argument( "-db", @@ -1871,7 +1881,40 @@ def main(): action="store_true", required=False, help="Whether to remove duplicated rows from the modified database for use with gget mutate (only for use with --download_cosmic).", - ) + ), + parser_cosmic.add_argument( + "--seq_id_column", + default="seq_ID", + type=str, + required=False, + help="Whether to remove duplicated rows from the modified database for use with gget mutate (only for use with --download_cosmic).", + ), + parser_cosmic.add_argument( + "--mutation_column", + default="mutation", + type=str, + required=False, + help="Whether to remove duplicated rows from the modified database for use with gget mutate (only for use with --download_cosmic).", + ), + parser_cosmic.add_argument( + "--mut_id_column", + default="mutation_id", + type=str, + required=False, + help="Whether to remove duplicated rows from the modified database for use with gget mutate (only for use with --download_cosmic).", + ), + parser_cosmic.add_argument( + "--email", + type=str, + required=False, + help="Email for COSMIC login. Helpful for avoiding required input upon running gget COSMIC. Default: None", + ), + parser_cosmic.add_argument( + "--password", + type=str, + required=False, + help="Password for COSMIC login. Helpful for avoiding required input upon running gget COSMIC, but password will be stored in plain text in the script. Default: None", + ), parser_cosmic.add_argument( "-o", "--out", @@ -1969,115 +2012,13 @@ def main(): required=False, help="Name of the column containing the IDs of each mutation in 'mutations'. Default: Same as 'mut_column'.", ) - parser_mutate.add_argument( - "-gtf", - "--gtf", - default=None, - type=str, - required=False, - help="Path to a .gtf file. When providing a genome fasta file as input for 'sequences', you can provide a .gtf file here and the input sequences will be defined according to the transcript boundaries, e.g. 'path/to/genome_annotation.gtf'.", - ) - parser_mutate.add_argument( - "-gtic", - "--gtf_transcript_id_column", - default=None, - type=str, - required=False, - help="Column name in the input 'mutations' file containing the transcript ID. In this case, column 'seq_id_column' should contain the chromosome number. Required when 'gtf' is provided.", - ) parser_mutate.add_argument( "-k", "--k", - default=30, - type=int, - required=False, - help="Length of sequences flanking the mutation. If k > total length of the sequence, the entire sequence will be kept.", - ) - parser_mutate.add_argument( - "-msl", - "--min_seq_len", default=None, type=int, required=False, - help="Minimum length of the mutant output sequence, e.g. 100. Mutant sequences smaller than this will be dropped.", - ) - parser_mutate.add_argument( - "-ma", - "--max_ambiguous", - default=None, - type=int, - required=False, - help="Maximum number of 'N' (or 'n') characters allowed in the output sequence, e.g. 10. Default: None (no ambiguous character filter will be applied).", - ) - parser_mutate.add_argument( - "-ofr", - "--optimize_flanking_regions", - default=False, - action="store_true", - required=False, - help="Removes nucleotides from either end of the mutant sequence to ensure (when possible) that the mutant sequence does not contain any k-mers also found in the wildtype/input sequence.", - ) - parser_mutate.add_argument( - "-rswk", - "--remove_seqs_with_wt_kmers", - default=False, - action="store_true", - required=False, - help="Removes output sequences where at least one k-mer is also present in the wildtype/input sequence in the same region. When used with `--optimize_flanking_regions`, only sequences for which a wildtpye kmer is still present after optimization will be removed.", - ) - parser_mutate.add_argument( - "-mio", - "--merge_identical_off", - default=True, - action="store_false", - required=False, - help="Do not merge identical mutant sequences in the output (by default, identical sequences will be merged by concatenating the sequence headers for all identical sequences).", - ) - parser_mutate.add_argument( - "-udf", - "--update_df", - default=False, - action="store_true", - required=False, - help="Updates the input `mutations` DataFrame to include additional columns with the mutation type, wildtype nucleotide sequence, and mutant nucleotide sequence (only valid if `mutations` is a .csv or .tsv file).", - ) - parser_mutate.add_argument( - "-udf_o", - "--update_df_out", - default=None, - type=str, - required=False, - help="Path to output csv file containing the updated DataFrame, e.g. 'path/to/mutations_updated.csv'. Only valid when used with `--update_df`. Default: None -> the new csv file will be saved in the same directory as the `mutations` DataFrame with appendix '_updated'.", - ) - parser_mutate.add_argument( - "--translate", - default=None, - action="store_true", - required=False, - help="Adds additional columns to the updated `mutations` DataFrame containing the wildtype and mutant amino acid sequences. Only valid when used with `--store_full_sequences`.", - ) - parser_mutate.add_argument( - "-ts", - "--translate_start", - default=None, - type=int_or_str, - required=False, - help="(int or str) The position in the input nucleotide sequence to start translating, e.g. 5. If a string is provided, it should correspond to a column name in `mutations` containing the open reading frame start positions for each sequence/mutation. Only valid when used with `--translate`. Default: translates from the beginning of each sequence.", - ) - parser_mutate.add_argument( - "--translate_end", - default=None, - type=int_or_str, - required=False, - help="(int or str) The position in the input nucleotide sequence to end translating, e.g. 35. If a string is provided, it should correspond to a column name in `mutations` containing the open reading frame end positions for each sequence/mutation. Only valid when used with `--translate`. Default: translates until the end of each sequence.", - ) - parser_mutate.add_argument( - "-sfs", - "--store_full_sequences", - default=False, - action="store_true", - required=False, - help="Includes the complete wildtype and mutant sequences in the updated `mutations` DataFrame (not just the sub-sequence with k-length flanks). Only valid when used with `--update_df`.", + help="Length of sequences flanking the mutation. If k is None or k > total length of the sequence, the entire sequence will be kept. Default: None", ) parser_mutate.add_argument( "-o", @@ -2366,7 +2307,8 @@ def main(): type=str, choices=["orthologs", "expression"], help="Type of information to be returned.", - required=True, + default="orthologs", + required=False, ) parser_bgee.add_argument( "-o", @@ -2395,6 +2337,32 @@ def main(): help="Does not print progress information.", ) + ## dataverse parser arguments + dataverse_desc = "Download datasets from the Dataverse repositories." + parser_dataverse = parent_subparsers.add_parser( + "dataverse", + parents=[parent], + description=dataverse_desc, + help=dataverse_desc, + add_help=True, + formatter_class=CustomHelpFormatter, + ) + parser_dataverse.add_argument( + "-o", + "--path", + type=str, + required=True, + help="Path to the directory the datasets will be saved in, e.g. 'path/to/directory'.", + ) + parser_dataverse.add_argument( + "-t", + "--table", + type=str, + default=None, + required=False, + help="File containing the dataset IDs to download, e.g. 'datasets.tsv'.", + ) + ### Define return values args = parent_parser.parse_args() @@ -2446,6 +2414,7 @@ def main(): "opentargets": parser_opentargets, "cbio": parser_cbio, "bgee": parser_bgee, + "dataverse": parser_dataverse, } if len(sys.argv) == 2: @@ -2628,23 +2597,10 @@ def main(): mutate_results = mutate( sequences=seqs, mutations=muts, - gtf=args.gtf, - gtf_transcript_id_column=args.gtf_transcript_id_column, k=args.k, mut_column=args.mut_column, mut_id_column=args.mut_id_column, seq_id_column=args.seq_id_column, - min_seq_len=args.min_seq_len, - max_ambiguous=args.max_ambiguous, - optimize_flanking_regions=args.optimize_flanking_regions, - remove_seqs_with_wt_kmers=args.remove_seqs_with_wt_kmers, - merge_identical=args.merge_identical_off, - update_df=args.update_df, - update_df_out=args.update_df_out, - store_full_sequences=args.store_full_sequences, - translate=args.translate, - translate_start=args.translate_start, - translate_end=args.translate_end, out=args.out, verbose=args.quiet, ) @@ -2794,6 +2750,7 @@ def main(): diamond_results = diamond( query=args.query, reference=args.reference, + translated=args.translated, diamond_db=args.diamond_db, sensitivity=args.sensitivity, threads=args.threads, @@ -3262,6 +3219,20 @@ def main(): else: print(json.dumps(pdb_results, ensure_ascii=False, indent=4)) + ## dataverse return + if args.command == "dataverse": + # Define separator based on file extension + if '.csv' in args.table: + sep = ',' + elif '.tsv' in args.table: + sep = '\t' + # Run gget dataverse function + dataverse( + df = args.table, + path = args.out, + sep = sep, + ) + ## opentargets return if args.command == "opentargets": flag_to_filter_id = { @@ -3367,3 +3338,18 @@ def main(): print( bgee_results.to_json(orient="records", force_ascii=False, indent=4) ) + + ## dataverse return + if args.command == "dataverse": + # Define separator based on file extension + if '.csv' in args.table: + sep = ',' + elif '.tsv' in args.table: + sep = '\t' + # Run gget dataverse function + dataverse( + df = args.table, + path = args.out, + sep = sep, + ) + \ No newline at end of file diff --git a/gget/utils.py b/gget/utils.py index 2483c3e1..5de7c1ed 100644 --- a/gget/utils.py +++ b/gget/utils.py @@ -4,6 +4,7 @@ # from requests.adapters import HTTPAdapter, Retry # import time import re +import sys import os import uuid import pandas as pd @@ -59,6 +60,15 @@ def set_up_logger(): logger = set_up_logger() +def print_sys(s): + """system print + + Args: + s (str): the string to print + """ + print(s, flush = True, file = sys.stderr) + + def flatten(xss): """ Function to flatten a list of lists. diff --git a/tests/fixtures.py b/tests/fixtures.py index de7ab515..0e13017c 100644 --- a/tests/fixtures.py +++ b/tests/fixtures.py @@ -1,5 +1,5 @@ # Latest Ensembl release for unittests -LATEST_ENS_RELEASE = 112 +LATEST_ENS_RELEASE = 113 # gget search species options for Ensembl release 106 SPECIES_OPTIONS = [ diff --git a/tests/fixtures/test_bgee.json b/tests/fixtures/test_bgee.json index 4e4ab266..1c8e1159 100644 --- a/tests/fixtures/test_bgee.json +++ b/tests/fixtures/test_bgee.json @@ -193,7 +193,6 @@ "test_bgee_orthologs": { "type": "assert_equal", "args": { - "type": "orthologs", "gene_id": "ENSOARG00000019163" }, "expected_result": [ @@ -699,4 +698,4 @@ }, "expected_result": "ValueError" } -} \ No newline at end of file +} diff --git a/tests/fixtures/test_blast.json b/tests/fixtures/test_blast.json index b1934828..4c2e3292 100644 --- a/tests/fixtures/test_blast.json +++ b/tests/fixtures/test_blast.json @@ -2,48 +2,24 @@ "test_blast_nt": { "type": "assert_equal", "args": { - "sequence": "ATACTCAGTCACACAAGCCATAGCAGGAAACAGCGAGCTTGCAGCCTCACCGACGAGTCTCAACTAAAAGGGACTCCCGGAGCTAGGGGTGGGGACTCGGCCTCACACAGTGAGTGCCGG", + "sequence": "MSKGEELFTGVVPILVELDGDVNGQKFSVSGEGEGDATYGKL", "limit": 1 }, "expected_result": [ [ - "Homo sapiens ATAC-STARR-seq lymphoblastoid active region 16974 (LOC129935398) on chromosome 2", - "Homo sapiens", - "human", - 9606, - 222, - 222, + "GFP deletion mutant [synthetic construct]", + "synthetic construct", + 32630, + 84.0, + 84.0, "100%", - 5.999999999999999e-54, + 1e-19, "100.00%", - 460, - "NG_168413.1" + 59, + "BAQ25552.1" ] ] }, - "test_blast_nt_json": { - "type": "assert_equal", - "args": { - "sequence": "ATACTCAGTCACACAAGCCATAGCAGGAAACAGCGAGCTTGCAGCCTCACCGACGAGTCTCAACTAAAAGGGACTCCCGGAGCTAGGGGTGGGGACTCGGCCTCACACAGTGAGTGCCGG", - "limit": 1, - "json": true - }, - "expected_result": [ - { - "Description": "Homo sapiens ATAC-STARR-seq lymphoblastoid active region 16974 (LOC129935398) on chromosome 2", - "Scientific Name": "Homo sapiens", - "Common Name": "human", - "Taxid": 9606, - "Max Score": 222, - "Total Score": 222, - "Query Cover": "100%", - "E value": 6e-54, - "Per. Ident": "100.00%", - "Acc. Len": 460, - "Accession": "NG_168413.1" - } - ] - }, "test_blast_bad_seq": { "type": "error", "args": { @@ -95,4 +71,4 @@ }, "expected_result": "ValueError" } -} \ No newline at end of file +} diff --git a/tests/fixtures/test_cbio.json b/tests/fixtures/test_cbio.json index 65ed613d..6bee468c 100644 --- a/tests/fixtures/test_cbio.json +++ b/tests/fixtures/test_cbio.json @@ -9,7 +9,7 @@ }, "expected_result": { "/tmp/test_cbio_download/msk_impact_2017/clinical_patient.txt": "08caa5754ffcae79b39eecad37edf201", - "/tmp/test_cbio_download/msk_impact_2017/clinical_sample.txt": "7550a0468bb410a2bdef42bd27c5ad32", + "/tmp/test_cbio_download/msk_impact_2017/clinical_sample.txt": "8d49c13c7ae2f4d0e2fbccb5ebf78316", "/tmp/test_cbio_download/msk_impact_2017/cna.txt": "e79f73232793853e808f69bd9d034125", "/tmp/test_cbio_download/msk_impact_2017/mutations.txt": "35264fd2f43492f09a15f15516fbe1b4", "/tmp/test_cbio_download/msk_impact_2017/sv.txt": "24ce260e03b417d47929f8fc34fc49a4" @@ -26,7 +26,7 @@ }, "expected_result": { "/tmp/test_cbio_download/msk_impact_2017/clinical_patient.txt": "08caa5754ffcae79b39eecad37edf201", - "/tmp/test_cbio_download/msk_impact_2017/clinical_sample.txt": "7550a0468bb410a2bdef42bd27c5ad32", + "/tmp/test_cbio_download/msk_impact_2017/clinical_sample.txt": "8d49c13c7ae2f4d0e2fbccb5ebf78316", "/tmp/test_cbio_download/msk_impact_2017/cna.txt": "e79f73232793853e808f69bd9d034125", "/tmp/test_cbio_download/msk_impact_2017/mutations.txt": "35264fd2f43492f09a15f15516fbe1b4", "/tmp/test_cbio_download/msk_impact_2017/sv.txt": "24ce260e03b417d47929f8fc34fc49a4", @@ -47,4 +47,4 @@ }, "expected_result": false } -} \ No newline at end of file +} diff --git a/tests/fixtures/test_cbio_search.json b/tests/fixtures/test_cbio_search.json index 7f724b24..6ff9d2e8 100644 --- a/tests/fixtures/test_cbio_search.json +++ b/tests/fixtures/test_cbio_search.json @@ -30,8 +30,9 @@ "ov_tcga_pan_can_atlas_2018", "ov_tcga_pub", "ovary_cptac_gdc", + "ovary_geomx_gray_foundation_2024", "scco_mskcc", "stes_tcga_pub" ] } -} \ No newline at end of file +} diff --git a/tests/fixtures/test_cosmic.json b/tests/fixtures/test_cosmic.json index 9d9e90b4..b3b18f89 100644 --- a/tests/fixtures/test_cosmic.json +++ b/tests/fixtures/test_cosmic.json @@ -1,247 +1,247 @@ { - "test_cosmic_defaults": { - "type": "assert_equal", - "args": { - "searchterm": "v600e" - }, - "expected_result": [ - [ - "BRAF", - "c.1799T>A", - "BRAF c.1799T>A, BRAF p.V600E, BRAF 1799T>A, BRAF V600E, BRAF COSV56056643, BRAF COSM476", - "y" - ], - [ - "BRAF", - "c.1799_1800del", - "BRAF c.1799_1800del, BRAF p.V600Efs*11, BRAF 1799_1800del, BRAF V600Efs*11, BRAF COSV56085831, BRAF COSM1168053", - "y" - ], - [ - "BRAF", - "c.1799_1800delinsAA", - "BRAF c.1799_1800delinsAA, BRAF p.V600E, BRAF 1799_1800delinsAA, BRAF V600E, BRAF COSV56059110, BRAF COSM475", - "y" - ], - [ - "BRAF", - "c.?", - "BRAF c.?, BRAF p.V600E, BRAF ?, BRAF V600E, BRAF COSV, BRAF COSM1131", - "y" - ], - [ - "DUSP27", - "c.1799T>A", - "DUSP27 c.1799T>A, DUSP27 p.V600E, DUSP27 1799T>A, DUSP27 V600E, DUSP27 COSV54807118, DUSP27 COSM358260", - "y" - ], - [ - "BAZ2A_ENST00000549884", - "c.1799T>A", - "BAZ2A_ENST00000549884 c.1799T>A, BAZ2A_ENST00000549884 p.V600E, BAZ2A_ENST00000549884 1799T>A, BAZ2A_ENST00000549884 V600E, BAZ2A_ENST00000549884 COSV51631953, BAZ2A_ENST00000549884 COSM6073024", - "n" - ], - [ - "BRAF_ENST00000496384", - "c.1799T>A", - "BRAF_ENST00000496384 c.1799T>A, BRAF_ENST00000496384 p.V600E, BRAF_ENST00000496384 1799T>A, BRAF_ENST00000496384 V600E, BRAF_ENST00000496384 COSV56056643, BRAF_ENST00000496384 COSM476", - "n" - ], - [ - "BRAF_ENST00000496384", - "c.1799_1800del", - "BRAF_ENST00000496384 c.1799_1800del, BRAF_ENST00000496384 p.V600Efs*11, BRAF_ENST00000496384 1799_1800del, BRAF_ENST00000496384 V600Efs*11, BRAF_ENST00000496384 COSV56085831, BRAF_ENST00000496384 COSM1168053", - "n" - ], - [ - "BRAF_ENST00000496384", - "c.1799_1800delinsAA", - "BRAF_ENST00000496384 c.1799_1800delinsAA, BRAF_ENST00000496384 p.V600E, BRAF_ENST00000496384 1799_1800delinsAA, BRAF_ENST00000496384 V600E, BRAF_ENST00000496384 COSV56059110, BRAF_ENST00000496384 COSM475", - "n" - ], - [ - "DUSP27_ENST00000271385", - "c.1799T>A", - "DUSP27_ENST00000271385 c.1799T>A, DUSP27_ENST00000271385 p.V600E, DUSP27_ENST00000271385 1799T>A, DUSP27_ENST00000271385 V600E, DUSP27_ENST00000271385 COSV54807118, DUSP27_ENST00000271385 COSM358260", - "n" - ], - [ - "DUSP27_ENST00000443333", - "c.1799T>A", - "DUSP27_ENST00000443333 c.1799T>A, DUSP27_ENST00000443333 p.V600E, DUSP27_ENST00000443333 1799T>A, DUSP27_ENST00000443333 V600E, DUSP27_ENST00000443333 COSV54807118, DUSP27_ENST00000443333 COSM358260", - "n" - ], - [ - "FMR1_ENST00000218200", - "c.1799T>A", - "FMR1_ENST00000218200 c.1799T>A, FMR1_ENST00000218200 p.V600E, FMR1_ENST00000218200 1799T>A, FMR1_ENST00000218200 V600E, FMR1_ENST00000218200 COSV54427979, FMR1_ENST00000218200 COSM756011", - "n" - ] - ] + "test_cosmic_defaults": { + "type": "assert_equal", + "args": { + "searchterm": "v600e" }, - "test_cosmic_limit_and_pubmet": { - "type": "assert_equal", - "args": { - "searchterm": "v600e", - "entity": "pubmed", - "limit": 2 - }, - "expected_result": [ - [ - "37546400", - "Advanced pulmonary sarcomatoid carcinoma patient harboring a braf lt sup gt v600e lt sup gt mutation responds to dabrafenib and trametinib a case report and literature review, an", - "Fang R,Gong J and Liao Z" - ], - [ - "21882184", - "Association of the braf v600e mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer a meta analysis, the", - "Kim TH,Park YJ,Lim JA,Ahn HY,Lee EK,Lee YJ,Kim KW,Hahn SK,Youn YK,Kim KH,Cho BY and Park do J" - ] - ] + "expected_result": [ + [ + "BRAF", + "c.1799T>A", + "BRAF c.1799T>A, BRAF p.V600E, BRAF 1799T>A, BRAF V600E, BRAF COSV56056643, BRAF COSM476", + "y" + ], + [ + "BRAF", + "c.1799_1800del", + "BRAF c.1799_1800del, BRAF p.V600Efs*11, BRAF 1799_1800del, BRAF V600Efs*11, BRAF COSV56085831, BRAF COSM1168053", + "y" + ], + [ + "BRAF", + "c.1799_1800delinsAA", + "BRAF c.1799_1800delinsAA, BRAF p.V600E, BRAF 1799_1800delinsAA, BRAF V600E, BRAF COSV56059110, BRAF COSM475", + "y" + ], + [ + "BRAF", + "c.?", + "BRAF c.?, BRAF p.V600E, BRAF ?, BRAF V600E, BRAF COSV, BRAF COSM1131", + "y" + ], + [ + "DUSP27", + "c.1799T>A", + "DUSP27 c.1799T>A, DUSP27 p.V600E, DUSP27 1799T>A, DUSP27 V600E, DUSP27 COSV54807118, DUSP27 COSM358260", + "y" + ], + [ + "BAZ2A_ENST00000549884", + "c.1799T>A", + "BAZ2A_ENST00000549884 c.1799T>A, BAZ2A_ENST00000549884 p.V600E, BAZ2A_ENST00000549884 1799T>A, BAZ2A_ENST00000549884 V600E, BAZ2A_ENST00000549884 COSV51631953, BAZ2A_ENST00000549884 COSM6073024", + "n" + ], + [ + "BRAF_ENST00000496384", + "c.1799T>A", + "BRAF_ENST00000496384 c.1799T>A, BRAF_ENST00000496384 p.V600E, BRAF_ENST00000496384 1799T>A, BRAF_ENST00000496384 V600E, BRAF_ENST00000496384 COSV56056643, BRAF_ENST00000496384 COSM476", + "n" + ], + [ + "BRAF_ENST00000496384", + "c.1799_1800del", + "BRAF_ENST00000496384 c.1799_1800del, BRAF_ENST00000496384 p.V600Efs*11, BRAF_ENST00000496384 1799_1800del, BRAF_ENST00000496384 V600Efs*11, BRAF_ENST00000496384 COSV56085831, BRAF_ENST00000496384 COSM1168053", + "n" + ], + [ + "BRAF_ENST00000496384", + "c.1799_1800delinsAA", + "BRAF_ENST00000496384 c.1799_1800delinsAA, BRAF_ENST00000496384 p.V600E, BRAF_ENST00000496384 1799_1800delinsAA, BRAF_ENST00000496384 V600E, BRAF_ENST00000496384 COSV56059110, BRAF_ENST00000496384 COSM475", + "n" + ], + [ + "DUSP27_ENST00000271385", + "c.1799T>A", + "DUSP27_ENST00000271385 c.1799T>A, DUSP27_ENST00000271385 p.V600E, DUSP27_ENST00000271385 1799T>A, DUSP27_ENST00000271385 V600E, DUSP27_ENST00000271385 COSV54807118, DUSP27_ENST00000271385 COSM358260", + "n" + ], + [ + "DUSP27_ENST00000443333", + "c.1799T>A", + "DUSP27_ENST00000443333 c.1799T>A, DUSP27_ENST00000443333 p.V600E, DUSP27_ENST00000443333 1799T>A, DUSP27_ENST00000443333 V600E, DUSP27_ENST00000443333 COSV54807118, DUSP27_ENST00000443333 COSM358260", + "n" + ], + [ + "FMR1_ENST00000218200", + "c.1799T>A", + "FMR1_ENST00000218200 c.1799T>A, FMR1_ENST00000218200 p.V600E, FMR1_ENST00000218200 1799T>A, FMR1_ENST00000218200 V600E, FMR1_ENST00000218200 COSV54427979, FMR1_ENST00000218200 COSM756011", + "n" + ] + ] + }, + "test_cosmic_limit_and_pubmet": { + "type": "assert_equal", + "args": { + "searchterm": "v600e", + "entity": "pubmed", + "limit": 2 }, - "test_cosmic_json_and_genes": { - "type": "assert_equal", - "args": { - "searchterm": "EGFR", - "entity": "genes", - "json": true - }, - "expected_result": [ - { - "Gene": "EGFR", - "Alternate IDs": "EGFR,ENST00000275493.6,EGFR,ENSP00000275493.2,Erlotinib,HKI-272,BIBW2992,Gefitinib,EGFR.html,NP_005219,NM_005228.3,ENSG00000146648.17,3236,131550,P00533,CCDS5514.1,1956,ERBB1,ERBB,COSG150", - "Tested samples": "212170", - "Simple Mutations": "32070", - "Fusions": "0", - "Coding Mutations": "32070" - }, - { - "Gene": "EGFR_ENST00000454757", - "Alternate IDs": "EGFR_ENST00000454757,ENST00000454757.6,EGFR,ENSP00000395243.3,EGFR.html,NP_001333828.1,ENSG00000146648.17,3236,131550,1956,ERBB1,ERBB,COSG454757", - "Tested samples": "212170", - "Simple Mutations": "10357", - "Fusions": "0", - "Coding Mutations": "10357" - }, - { - "Gene": "EGFR_ENST00000455089", - "Alternate IDs": "EGFR_ENST00000455089,ENST00000455089.5,EGFR,ENSP00000415559.1,EGFR.html,NP_001333826.1,NM_001346897.1,ENSG00000146648.17,3236,131550,1956,ERBB1,ERBB,COSG455089", - "Tested samples": "212170", - "Simple Mutations": "10101", - "Fusions": "0", - "Coding Mutations": "10101" - }, - { - "Gene": "EGFR_ENST00000638463", - "Alternate IDs": "EGFR_ENST00000638463,ENST00000638463.1,EGFR,ENSP00000492462.1,EGFR.html,ENSG00000146648.17,3236,131550,1956,ERBB1,ERBB,COSG638463", - "Tested samples": "212169", - "Simple Mutations": "9518", - "Fusions": "0", - "Coding Mutations": "9518" - }, - { - "Gene": "EGFR_ENST00000344576", - "Alternate IDs": "EGFR_ENST00000344576,ENST00000344576.6,EGFR,ENSP00000345973.2,EGFR.html,NP_958441,NM_201284.1,ENSG00000146648.17,3236,131550,P00533,CCDS5515.1,1956,ERBB1,ERBB,COSG90589", - "Tested samples": "212167", - "Simple Mutations": "2401", - "Fusions": "0", - "Coding Mutations": "2401" - }, - { - "Gene": "EGFR_ENST00000342916", - "Alternate IDs": "EGFR_ENST00000342916,ENST00000342916.7,EGFR,ENSP00000342376.3,EGFR.html,NP_958439,NM_201282.1,ENSG00000146648.17,3236,131550,P00533,CCDS5516.1,1956,ERBB1,ERBB,COSG107618", - "Tested samples": "212167", - "Simple Mutations": "2265", - "Fusions": "0", - "Coding Mutations": "2265" - }, - { - "Gene": "EGFR_ENST00000420316", - "Alternate IDs": "EGFR_ENST00000420316,ENST00000420316.6,EGFR,ENSP00000413843.2,EGFR.html,NP_958440,NM_201283.1,ENSG00000146648.17,3236,131550,P00533,CCDS47587.1,1956,ERBB1,ERBB,COSG420316", - "Tested samples": "212164", - "Simple Mutations": "1754", - "Fusions": "0", - "Coding Mutations": "1754" - }, - { - "Gene": "RHBDF1", - "Alternate IDs": "RHBDF1,ENST00000262316.10,RHBDF1,ENSP00000262316.5,NP_071895,NM_022450.3,ENSG00000007384.15,20561,614403,Q96CC6,CCDS32344.1,64285,iRhom1,FLJ2235,EGFR-RS,Dist1,C16orf8,COSG67901", - "Tested samples": "46013", - "Simple Mutations": "580", - "Fusions": "0", - "Coding Mutations": "580" - } - ] + "expected_result": [ + [ + "37546400", + "Advanced pulmonary sarcomatoid carcinoma patient harboring a braf lt sup gt v600e lt sup gt mutation responds to dabrafenib and trametinib a case report and literature review, an", + "Fang R,Gong J and Liao Z" + ], + [ + "21882184", + "Association of the braf v600e mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer a meta analysis, the", + "Kim TH,Park YJ,Lim JA,Ahn HY,Lee EK,Lee YJ,Kim KW,Hahn SK,Youn YK,Kim KH,Cho BY and Park do J" + ] + ] + }, + "test_cosmic_json_and_genes": { + "type": "assert_equal", + "args": { + "searchterm": "EGFR", + "entity": "genes", + "json": true }, - "test_cosmic_samples": { - "type": "assert_equal", - "args": { - "searchterm": "EGFR", - "entity": "samples" - }, - "expected_result": [ - [ - "P1_Pre-RAFi_EGFRi", - "Large intestine, carcinoma, adenocarcinoma", - "1224", - "1179", - "0", - "0" - ] - ] + "expected_result": [ + { + "Gene": "EGFR", + "Alternate IDs": "EGFR,ENST00000275493.6,EGFR,ENSP00000275493.2,Erlotinib,HKI-272,BIBW2992,Gefitinib,EGFR.html,NP_005219,NM_005228.3,ENSG00000146648.17,3236,131550,P00533,CCDS5514.1,1956,ERBB1,ERBB,COSG150", + "Tested samples": "215828", + "Simple Mutations": "32442", + "Fusions": "0", + "Coding Mutations": "32442" + }, + { + "Gene": "EGFR_ENST00000454757", + "Alternate IDs": "EGFR_ENST00000454757,ENST00000454757.6,EGFR,ENSP00000395243.3,EGFR.html,NP_001333828.1,ENSG00000146648.17,3236,131550,1956,ERBB1,ERBB,COSG454757", + "Tested samples": "215828", + "Simple Mutations": "10498", + "Fusions": "0", + "Coding Mutations": "10498" + }, + { + "Gene": "EGFR_ENST00000455089", + "Alternate IDs": "EGFR_ENST00000455089,ENST00000455089.5,EGFR,ENSP00000415559.1,EGFR.html,NP_001333826.1,NM_001346897.1,ENSG00000146648.17,3236,131550,1956,ERBB1,ERBB,COSG455089", + "Tested samples": "215828", + "Simple Mutations": "10240", + "Fusions": "0", + "Coding Mutations": "10240" + }, + { + "Gene": "EGFR_ENST00000638463", + "Alternate IDs": "EGFR_ENST00000638463,ENST00000638463.1,EGFR,ENSP00000492462.1,EGFR.html,ENSG00000146648.17,3236,131550,1956,ERBB1,ERBB,COSG638463", + "Tested samples": "215827", + "Simple Mutations": "9649", + "Fusions": "0", + "Coding Mutations": "9649" + }, + { + "Gene": "EGFR_ENST00000344576", + "Alternate IDs": "EGFR_ENST00000344576,ENST00000344576.6,EGFR,ENSP00000345973.2,EGFR.html,NP_958441,NM_201284.1,ENSG00000146648.17,3236,131550,P00533,CCDS5515.1,1956,ERBB1,ERBB,COSG90589", + "Tested samples": "215825", + "Simple Mutations": "2427", + "Fusions": "0", + "Coding Mutations": "2427" + }, + { + "Gene": "EGFR_ENST00000342916", + "Alternate IDs": "EGFR_ENST00000342916,ENST00000342916.7,EGFR,ENSP00000342376.3,EGFR.html,NP_958439,NM_201282.1,ENSG00000146648.17,3236,131550,P00533,CCDS5516.1,1956,ERBB1,ERBB,COSG107618", + "Tested samples": "215825", + "Simple Mutations": "2288", + "Fusions": "0", + "Coding Mutations": "2288" + }, + { + "Gene": "EGFR_ENST00000420316", + "Alternate IDs": "EGFR_ENST00000420316,ENST00000420316.6,EGFR,ENSP00000413843.2,EGFR.html,NP_958440,NM_201283.1,ENSG00000146648.17,3236,131550,P00533,CCDS47587.1,1956,ERBB1,ERBB,COSG420316", + "Tested samples": "215822", + "Simple Mutations": "1773", + "Fusions": "0", + "Coding Mutations": "1773" + }, + { + "Gene": "RHBDF1", + "Alternate IDs": "RHBDF1,ENST00000262316.10,RHBDF1,ENSP00000262316.5,NP_071895,NM_022450.3,ENSG00000007384.15,20561,614403,Q96CC6,CCDS32344.1,64285,iRhom1,FLJ2235,EGFR-RS,Dist1,C16orf8,COSG67901", + "Tested samples": "49123", + "Simple Mutations": "589", + "Fusions": "0", + "Coding Mutations": "589" + } + ] + }, + "test_cosmic_samples": { + "type": "assert_equal", + "args": { + "searchterm": "EGFR", + "entity": "samples" }, - "test_cosmic_studies": { - "type": "assert_equal", - "args": { - "searchterm": "THCA-SA", - "entity": "studies" - }, - "expected_result": [ - [ - "589", - "THCA-SA", - "ICGC(THCA-SA) : Thyroid Cancer - SA" - ] - ] + "expected_result": [ + [ + "P1_Pre-RAFi_EGFRi", + "Large intestine, carcinoma, adenocarcinoma", + "1224", + "1179", + "0", + "0" + ] + ] + }, + "test_cosmic_studies": { + "type": "assert_equal", + "args": { + "searchterm": "THCA-SA", + "entity": "studies" }, - "test_cosmic_cancer": { - "type": "assert_equal", - "args": { - "searchterm": "prostate", - "entity": "cancer", - "limit": 2 - }, - "expected_result": [ - [ - "haematopoietic and lymphoid tissue,lymphoid neoplasm", - "haematopoietic and lymphoid tissue,lymphoid neoplasm (prostate,lymphoma)", - "117534", - "1027628" - ], - [ - "prostate,carcinoma", - "prostate,carcinoma (prostate,neoplasm)", - "24201", - "793069" - ] - ] + "expected_result": [ + [ + "589", + "THCA-SA", + "ICGC(THCA-SA) : Thyroid Cancer - SA" + ] + ] + }, + "test_cosmic_cancer": { + "type": "assert_equal", + "args": { + "searchterm": "prostate", + "entity": "cancer", + "limit": 2 }, - "test_cosmic_tumour": { - "type": "assert_equal", - "args": { - "searchterm": "prostate", - "entity": "tumour_site", - "limit": 2 - }, - "expected_result": [ - [ - "prostate", - "24901", - "56007", - "2931819", - "2747", - "87043" - ] - ] - } -} \ No newline at end of file + "expected_result": [ + [ + "haematopoietic and lymphoid tissue,lymphoid neoplasm", + "haematopoietic and lymphoid tissue,lymphoid neoplasm (prostate,lymphoma)", + "117882", + "1041146" + ], + [ + "prostate,carcinoma", + "prostate,carcinoma (prostate,neoplasm)", + "24634", + "794299" + ] + ] + }, + "test_cosmic_tumour": { + "type": "assert_equal", + "args": { + "searchterm": "prostate", + "entity": "tumour_site", + "limit": 2 + }, + "expected_result": [ + [ + "prostate", + "25334", + "56007", + "2938196", + "2747", + "87143" + ] + ] + } +} diff --git a/tests/fixtures/test_info.json b/tests/fixtures/test_info.json index e1248501..f0230488 100644 --- a/tests/fixtures/test_info.json +++ b/tests/fixtures/test_info.json @@ -239,7 +239,7 @@ "Guanine nucleotide-binding protein G(i) subunit alpha-3", "G protein subunit alpha i3 [Source:MGI Symbol;Acc:MGI:95773]", "Heterotrimeric guanine nucleotide-binding proteins (G proteins) function as transducers downstream of G protein-coupled receptors (GPCRs) in numerous signaling cascades. The alpha chain contains the guanine nucleotide binding site and alternates between an active, GTP-bound state and an inactive, GDP-bound state. Signaling by an activated GPCR promotes GDP release and GTP binding. The alpha subunit has a low GTPase activity that converts bound GTP to GDP, thereby terminating the signal. Both GDP release and GTP hydrolysis are modulated by numerous regulatory proteins. Signaling is mediated via effector proteins, such as adenylate cyclase. Inhibits adenylate cyclase activity, leading to decreased intracellular cAMP levels. Stimulates the activity of receptor-regulated K(+) channels. The active GTP-bound form prevents the association of RGS14 with centrosomes and is required for the translocation of RGS14 from the cytoplasm to the plasma membrane. May play a role in cell division", - "Predicted to enable several functions, including G-protein beta/gamma-subunit complex binding activity; GDP binding activity; and GTPase activating protein binding activity. Predicted to be involved in several processes, including positive regulation of NAD(P)H oxidase activity; positive regulation of superoxide anion generation; and positive regulation of vascular associated smooth muscle cell proliferation. Predicted to act upstream of or within G protein-coupled receptor signaling pathway. Located in Golgi apparatus. Is expressed in early conceptus; inner ear; and oocyte. Orthologous to human GNAI3 (G protein subunit alpha i3). [provided by Alliance of Genome Resources, Apr 2022]", + "Predicted to enable several functions, including G-protein beta/gamma-subunit complex binding activity; GDP binding activity; and GTPase activating protein binding activity. Predicted to be involved in several processes, including G protein-coupled receptor signaling pathway; positive regulation of superoxide anion generation; and positive regulation of vascular associated smooth muscle cell proliferation. Predicted to act upstream of or within G protein-coupled receptor signaling pathway. Located in Golgi apparatus. Is expressed in early conceptus; inner ear; and oocyte. Orthologous to human GNAI3 (G protein subunit alpha i3). [provided by Alliance of Genome Resources, Dec 2024]", [ "Cytoplasm", "Cell membrane", @@ -1151,4 +1151,4 @@ }, "expected_result": null } -} \ No newline at end of file +} diff --git a/tests/fixtures/test_mutate.json b/tests/fixtures/test_mutate.json index e311a66b..1fa8c754 100644 --- a/tests/fixtures/test_mutate.json +++ b/tests/fixtures/test_mutate.json @@ -4,7 +4,7 @@ "args": { "sequences": "