+
diff --git a/installation_guide.md b/installation_guide.md
index be7c8de..15a17a1 100644
--- a/installation_guide.md
+++ b/installation_guide.md
@@ -16,6 +16,9 @@ This guide covers how to install and update XMapTools 4.5 and future releases on
- [Update Only (Windows)](#update-only-windows)
- [Show Information (Windows)](#show-information-windows)
- [Troubleshooting](#troubleshooting)
+ - [XMapTools suddenly not opening (Windows)](#xmaptools-suddenly-not-opening-windows)
+ - [Unfreeze XMapTools in case of error](#unfreeze-xmaptools-in-case-of-error)
+ - [Uninstalling XMapTools (Windows)](#uninstalling-xmaptools-windows)
## Prerequisites
@@ -236,6 +239,25 @@ Then re-run the installation or update command.
Make sure you are running PowerShell **as Administrator**. Right-click the PowerShell icon and select *Run as Administrator*.
+### XMapTools suddenly not opening (Windows)
+
+If XMapTools was working previously and suddenly stops opening, this can be caused by a corrupted MATLAB Runtime installation or a Windows update that modified system paths. Try the following:
+
+1. Perform a full reinstallation using the `--install` flag
+2. If the issue persists, uninstall the MATLAB Runtime (see [Delete Old Runtime Libraries](#delete-old-runtime-libraries)) and reinstall
+
+### Unfreeze XMapTools in case of error
+
+If XMapTools becomes unresponsive during an operation, a dialog box may appear with **OK** and **Cancel** buttons. Click **Cancel** to cancel the current operation and unfreeze XMapTools. Clicking **OK** may restart the operation and keep XMapTools frozen.
+
+### Uninstalling XMapTools (Windows)
+
+To fully uninstall XMapTools on Windows:
+
+1. Open **Settings → Apps & Features** (or **Control Panel → Programs and Features**)
+2. Find **XMapTools** in the list and click **Uninstall**
+3. Optionally, uninstall the **MATLAB Runtime** from the same list if no other MATLAB-based applications require it
+
---
## Quick Reference
diff --git a/public/assets/Logo_2025_square_512.png b/public/assets/Logo_2025_square_512.png
new file mode 100644
index 0000000..bd13bf4
Binary files /dev/null and b/public/assets/Logo_2025_square_512.png differ
diff --git a/src/assets/Logo_2025_square_512.png b/src/assets/Logo_2025_square_512.png
new file mode 100644
index 0000000..bd13bf4
Binary files /dev/null and b/src/assets/Logo_2025_square_512.png differ
diff --git a/tutorials.md b/tutorials.md
index 310df84..ac8582e 100644
--- a/tutorials.md
+++ b/tutorials.md
@@ -9,9 +9,13 @@ Follow the steps in this tutorial to familiarise yourself with the EPMA data red
A video tutorial is also available on YouTube and Bilibili:
-
+
+
+
+
+Located in China? Watch this video on Bilibili
+
-
### Dataset description
@@ -52,16 +56,17 @@ When the program is ready, a dialog box opens; pick the working directory __Tuto
### Import maps
-1. In the first workspace "PROJECT & IMPORT", click on the "Import" button (Import Maps) to open the Import Tool module.
+
+1. In the first workspace "PROJECT & IMPORT", click on the "Import" button (Import Maps) to open the Import Tool module.
2. Select all the map files to be imported and click on the button "Open". Note that you can select all the text files available in the folder and XMapTools will find out which ones can be imported as maps.
3. In the tab "Corrections", change the value of dwell time to 120 ms.
4. Press the "Import Data" button.
### X-ray data visualization (Part 1)
-1. Quickly go through the maps and use the auto-contrast option or the live frequency polygon (see figure below) to adjust the lower and upper limits of the color bar. Note that the color bar settings are available in the "Options" workspace.
+1. Quickly go through the maps and use the auto-contrast button or the live frequency polygon (see figure below) to adjust the lower and upper limits of the color bar. Note that the color bar settings are available in the "Options" workspace.
2. Eliminate the maps which are not needed because they do not contain any important information (Ce, Cr, Ni, S). To eliminate a map, select the map in the primary tree menu, then right click on the name and select "Delete".
-3. Save your project using either the "Save" button available in the "PROJECT & IMPORT" workspace or in the menu "File" > "Save Project".
+3. Save your project using either the "Save" button available in the "PROJECT & IMPORT" workspace or in the menu "File" > "Save Project".
@@ -72,12 +77,12 @@ Classification is the step used to create a classified image in which each pixel
Additional information about the classification in XMapTools can be found in : Lanari, P., Tedeschi, M., (2025). Chemical map classification in XMapTools. _Applied Computing and Geosciences_, __25__, 100230 [Download].
1. Set the active workspace to CLASSIFY.
-2. Create a new training set. In the secondary tree menu, select the category "Training Set (Classification)" and press the button "Add" available in the CLASSIFY workspace to add phase definitions. Select in the list the phases: Biotite, Garnet, Plagioclase, Quartz; then click "Apply".
+2. Create a new training set. In the secondary tree menu, select the category "Training Set (Classification)" and press the "Add" button available in the CLASSIFY workspace to add phase definitions. Select in the list the phases: Biotite, Garnet, Plagioclase, Quartz; then click "Apply".
3. Add training data for each phase:
- Select a training class in the training set.
- - Adjust display (optional): select an element to be display and eventually use the zoom and pan features to adjust the field of view.
+ - Adjust display (optional): select an element to be display and eventually use the zoom and pan buttons to adjust the field of view.
- Select a ROI type in the dropdown menu among "Rectangle ROI", "Polygon ROI", "Ellipse ROI" and "Circle ROI".
- - Finally, press the "Add a ROI" button and immediately click and drag (or click for polygon) over the map to create the ROI. How to draw a ROI in XMapTools:
+ - Finally, press the "Add a ROI" button and immediately click and drag (or click for polygon) over the map to create the ROI. How to draw a ROI in XMapTools:
- Rectangle: click to select the first corner and drag the mouse to the opposite corner defining a rectangle.
- Polygon: click successively on the image to draw a polygon; right-click or double-click to validate and close automatically the shape.
- Ellipse: click to select the first point and drag the mouse to a second point defining the long axis of an ellipse.
@@ -87,13 +92,13 @@ Additional information about the classification in XMapTools can be found in : L
6. Add new phase definitions for sillimanite and sudoite (low temperature chlorite here replacing cordierite). Add training data for each phase (see step 3 above).
7. Restore the view using either the button "Reset Zoom & Pan" or the button "Restore view" available near the top-right corner of the main figure.
8. In classification parameters:
- - Add all maps in the list for classification. Select an element to display in the primary tree menu and use the "Add all" button to add all maps at once.
- - Eliminate La from the list; select La_EDS in the primary tree menu and press the button "Take La_EDS out".
- - You can calculate PCA maps using the "Generate Maps of the Principal Component" button. The maps of principal components are listed in the category "Other" available in the primary tree menu.
+ - Add all maps in the list for classification. Select an element to display in the primary tree menu and use the "Add all" button to add all maps at once.
+ - Eliminate La from the list; select La_EDS in the primary tree menu and press the "Take La_EDS out" button.
+ - You can calculate PCA maps using the "Generate Maps of the Principal Component" button. The maps of principal components are listed in the category "Other" available in the primary tree menu.
- Select the data type "Maps" (other data types are described in Lanari & Tedeschi 2025).
- Select the map scaling method "Robust"; select the option "reproducibility" and set the seed value to 1.
9. Select the algorithm "Random Forest" and set the option corresponding to the maximum number of trees to 50.
-10. Select the phase definition object "PhaseDef_1" in the secondary tree menu and press the button "Classify".
+10. Select the phase definition object "PhaseDef_1" in the secondary tree menu and press the "Classify" button.
11. Select a mask file to display the mineral map.
12. Check the results of the classification (for Random Forest):
- Are all training pixels classified in the true class? If not, the training set should be adjusted accordingly.
@@ -107,18 +112,18 @@ Additional information about the classification in XMapTools can be found in : L
1. Select a mask file.
2. Select the ROI shape "rectangle" using the dropdown menu in "Mask analysis and visualization"
-3. Press the "Add a ROI to export mask modes" button and immediately click and drag over the map to create the ROI.
+3. Press the "Add a ROI to export mask modes" button and immediately click and drag over the map to create the ROI.
4. Adjust the shape to obtain the modes of the entire map. __If the editing mode is disabled, press once the "Ctrl" key__ on your keyboard and try again editing the shape of the ROI.
-5. Save the modes using the button save in the composition tab on the right.
-6. Note that you can also copy the data to the clipboard using the button copy. Paste the data in a spreadsheet software program.
-7. Eliminate the ROI using the function "Reset ROI".
+5. Save the modes using the save button in the composition tab on the right.
+6. Note that you can also copy the data to the clipboard using the copy button. Paste the data in a spreadsheet software program.
+7. Eliminate the ROI using the "Reset ROI" button.
8. For comparaison purposes (don't use that method for publications!), calculate pseudo modes using the point counting method:
- Set the number of points to 500.
- Unselect the Monte-Carlo option.
- - Extract several successive sets of pseudo modes using the point counting method by clicking on the "Generate pseudo-modes" button and copy the results in a spreadsheet software program. _Note: now you know why this method should not be used._
+ - Extract several successive sets of pseudo modes using the point counting method by clicking on the "Generate pseudo-modes" button and copy the results in a spreadsheet software program. _Note: now you know why this method should not be used._
9. Calculate pseudo modes using the point counting method and Monte Carlo to calculate uncertainties on the results (_note: what is never done with point counting_):
- Set the number of points to 300.
- - Select the Monte-Carlo optionExtract pseudo modes and uncertainties using the "Generate pseudo-modes" button and copy the results in a spreadsheet software program.
+ - Select the Monte-Carlo optionExtract pseudo modes and uncertainties using the "Generate pseudo-modes" button and copy the results in a spreadsheet software program.
- You can repeat this calculation for several number of points (e.g. 500, 750, 1000) and copy the results in a spreadsheet software program.
### Analytical standardisation
@@ -134,18 +139,18 @@ The calibration method used by XMapTools is described in Lanari, P., Vho, A., Bo
1. Set the active workspace to "CALIBRATE".
2. Display an intensity map showing a maximum of minerals (e.g. Al).
3. Select the option "Import standards.txt" in "EPMA STANDARD DATA".
-4. Click on the "Import" button. Note that the color of the spot labels can be changed in the options of the "Project & Import" workspace.
-5. Select the element Mg to estimate the quality of the position. The diagrams on the right show correlation maps with the center of the image being the position of the spots on the map. In this case the original position is good and there is no need to change it. (Optional) you can try to shift the spot position in the correlation plot of the element and then to press "apply". To restore the original position, you can simply load the standard data again using the import function.
+4. Click on the "Import" button. Note that the color of the spot labels can be changed in the options of the "Project & Import" workspace.
+5. Select the element Mg to estimate the quality of the position. The diagrams on the right show correlation maps with the center of the image being the position of the spots on the map. In this case the original position is good and there is no need to change it. (Optional) you can try to shift the spot position in the correlation plot of the element and then to press "apply" . To restore the original position, you can simply load the standard data again using the import button.
6. Add two additional internal standard values for quartz (not measured):
- Display the silicon map by selecting Si in the primary tree menu.
- Select the category "Standards (Spots)" in the secondary tree menu.
- Adjust the view using zoom and pan (optional)
- - Press the "Add" button available in the "CALIBRATE" workspace select a pixel of quartz by clicking on the main image.
+ - Press the "Add" button available in the "CALIBRATE" workspace select a pixel of quartz by clicking on the main image.
- Set the composition to 100 wt% of SiO2 in the table available in the "Standard" tab.
- Note that the new standard is listed at the bottom of the secondary tree menu under "Standards (Spots)". Selecting this internal standard display the location and the composition.
7. Calibrate the intensity element maps into maps of wt% of oxide:
- Select a mask file in the secondary tree menu.
- - In the "CALIBRATE" workspace, press the button "Calibrate" to open the Calibration assistant for EPMA data.
+ - In the "CALIBRATE" workspace, press the "Calibrate" button to open the Calibration assistant for EPMA data.
- Check the calibration curves by displaying the curve for each element of each mineral.
- Use the button "Adjust (Manual)" to modify the calibration curve in case the calibration algorithm failed to set a proper calibration curveSelect the option "Apply to all masks".
- Press the button "Apply Standardization" to calibrate all maps and send the data to XMapTools.
@@ -159,19 +164,19 @@ _Note: The video tutorial (see above) provides more tricks and explanations abou
1. We need first to create a density map for the mask file used for calibration:
- Select a mask file in the secondary tree menu.
- - In the "CALIBRATE" workspace, press the "Density" button to automatically generate a density map.
+ - In the "CALIBRATE" workspace, press the "Density" button to automatically generate a density map.
- Enter average density values for each phase. Note that average values are automatically proposed if the mineral name used is available in the XMapTools database.
- You can display the density map in that is listed under the category "Other" in the primary tree menu.
2. Display a merged map by selecting an oxide in the category "Merged" of the primary tree menu.
-3. Select "Rectangle ROI", then press the "Add ROI" button and immediately click and drag over the map to create the ROI.
+3. Select "Rectangle ROI", then press the "Add ROI" button and immediately click and drag over the map to create the ROI.
4. For this exercise, reshape the ROI to about ¾ of the map surface. If the editing mode is disabled, press once the "Ctrl" key and try again editing the shape of the ROI.
5. Save the local bulk composition.
-6. Set the number of simulations to 100 and the shift value expressed in number of pixels to 20 and press the "Calculate the uncertainties using Monte Carlo" button.
+6. Set the number of simulations to 100 and the shift value expressed in number of pixels to 20 and press the "Calculate the uncertainties using Monte Carlo" button.
7. Save the results and the figure (File > Save As…).
8. Change the ROI to cover the entire map and export the new bulk composition. Compare the composition with the one previously obtained for the smaller domain.
9. Create a new merged map "garnet-excluded" containing only pixel compositions of biotite, plagioclase, quartz, sillimanite and sudoite and export the local bulk composition:
- Select the item "Biotite" in the category "Quanti" of the primary tree menu.
- - Press the "Merge" button.
+ - Press the "Merge" button.
- Select all mineral except Garnet and press the "Apply" button.
- Rename the merged map into "Merged_noGrt".
- Export a local bulk composition of the entire area.
@@ -182,9 +187,9 @@ The method to extract local bulk composition is described in Lanari, P., & Engi,
1. Set the active workspace to "FUNCTIONS".
2. In "Normalization & Structural Formula", select "Function", "Biotite" and "Bt (SF, 11-Ox. basis)".
-3. Display the function description by clicking on the yellow help button.
+3. Display the function description by clicking on the yellow help button.
4. Select "Biotite" in the category "Quanti" of the primary tree menu
-5. Press the "Apply" button to calculate the maps of structural formula for biotite.
+5. Press the "Apply" button to calculate the maps of structural formula for biotite.
6. Display the maps of structural formula "Biotite Bt (SF, 11-Ox. basis)" available in the category "Results" of the primary tree menu.
7. Generate an image showing the maps of Si, Ti and XMg in biotite:
- In the menu, select in the menu: Image > Multi-Selection Mode.
@@ -206,7 +211,7 @@ The method to extract local bulk composition is described in Lanari, P., & Engi,
- Select any result in "Biotite Bt (SF, 11-Ox. basis)".
- In the menu, select: Modules > Generator.
- Type the following equation in the field that calculates Mg (apfu): "Mg = Mg_M1 + Mg_M2".
- - Press the "Generate" button.
+ - Press the "Generate" button.
- Activate the multi-selection mode.
- Select the maps Mg in garnet and Mg in biotite; both are expressed in apfu.
- In the menu "Image", select the option "Add Multi-Layer Image (shared-scale)".
@@ -215,7 +220,7 @@ The method to extract local bulk composition is described in Lanari, P., & Engi,
14. Calculate temperature maps using Ti-in-biotite thermometry:
- In the FUNCTIONS workspace, activate the "Map mode" in the section "Thermobarometry and other methods", select "Biotite" and "T.Bt (all calibrations)".
- Select "Biotite" in the category "Quanti".
- - Press the "Apply" button available in the section "Thermobarometry and other methods".
+ - Press the "Apply" button available in the section "Thermobarometry and other methods".
- Set the pressure to 6 kbar (0.6 GPa).
- Display the map T_H05.
- Save a single image containing the temperature map T_H05.
@@ -223,13 +228,13 @@ The method to extract local bulk composition is described in Lanari, P., & Engi,
- Select the merge map "Merged_RandomForest".
- Adjust the view using zoom and pan (optional).
- In the workspace "FUNCTIONS", activate the "Multi-equilibrium" mode in the section "Thermobarometry and other methods", select "Biotite" or "Garnet" and "T.Bt (all calibrations)".
- - Display the map FeOSelect "Circle ROI", then press the "Add ROI" button.
+ - Display the map FeOSelect "Circle ROI", then press the "Add ROI" button.
- Set the pressure to 6 kbar (0.6 GPa).
- Select a ROI for garnet and then a ROI for biotite.
- - Results are displayed in a table for all calibrations. Check the help function to obtain the references.
- - Save the results using the "Save" button available above the table.
+ - Results are displayed in a table for all calibrations. Check the help function to obtain the references.
+ - Save the results using the "Save" button available above the table.
- Adjust the position of the ROI and extract the new temperature.
- - Click on the "Reset ROI" button.
+ - Click on the "Reset ROI" button.
### Sampling tools and chemical diagrams
@@ -245,7 +250,7 @@ The method to extract local bulk composition is described in Lanari, P., & Engi,
- In the menu, select Sampling > Strip and click on the image to create rectangle.
- Adjust the rotation and the position of the rectangle to sample from rim to core of a garnet grain.
- Save the results and compare the profile obtained with the ones obtained using the transect tool.
- - Reset ROI using the button.
+ - Reset ROI using the button.
3. Generate a binary plot for biotite:
- Select any map of the structural formula of biotite.
- In the menu select Modules > Data Vizualisation.
diff --git a/videos.md b/videos.md
index 127b048..f13d9bd 100644
--- a/videos.md
+++ b/videos.md
@@ -1,3 +1,44 @@
# Videos
-Links to XMapTools videos
+Watch our videos to find out more and learn how to use XMapTools.
+
+## Table of contents
+
+- [LA-ICP-MS data reduction in XMapTools 4.3](#la-icp-ms-data-reduction-in-xmaptools-4-3)
+- [Introduction talk to XMapTools 4 for EPMA](#introduction-talk-to-xmaptools-4-for-epma)
+- [XMapTools Course 1 – How to get started (EPMA)?](#xmaptools-course-1-–-how-to-get-started-epma)
+- [XMapTools 4 Special Event – December 2021](#xmaptools-4-special-event-–-december-2021)
+
+
+## LA-ICP-MS data reduction in XMapTools 4.3
+
+

 ### Dataset description
@@ -52,16 +56,17 @@ When the program is ready, a dialog box opens; pick the working directory __Tuto
### Import maps
-1. In the first workspace "PROJECT & IMPORT", click on the "Import" button (Import Maps) to open the Import Tool module.
### Dataset description
@@ -52,16 +56,17 @@ When the program is ready, a dialog box opens; pick the working directory __Tuto
### Import maps
-1. In the first workspace "PROJECT & IMPORT", click on the "Import" button (Import Maps) to open the Import Tool module. 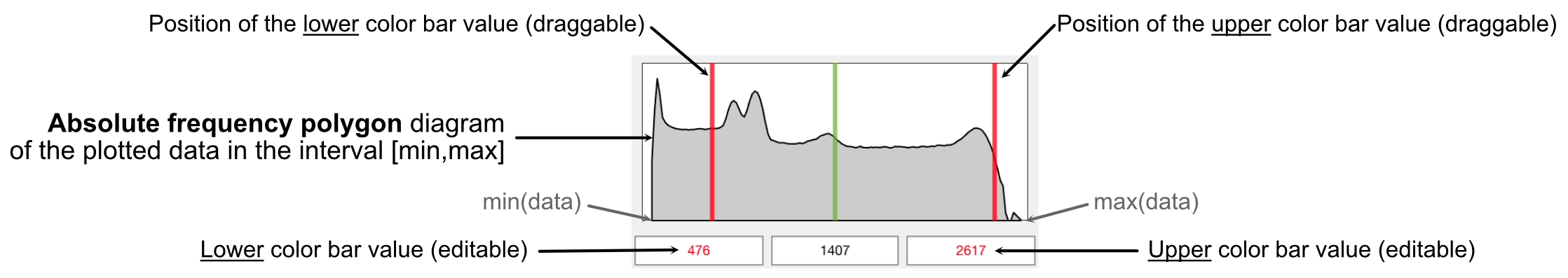 @@ -72,12 +77,12 @@ Classification is the step used to create a classified image in which each pixel
Additional information about the classification in XMapTools can be found in : Lanari, P., Tedeschi, M., (2025). Chemical map classification in XMapTools. _Applied Computing and Geosciences_, __25__, 100230 [Download].
1. Set the active workspace to CLASSIFY.
-2. Create a new training set. In the secondary tree menu, select the category "Training Set (Classification)" and press the button "Add" available in the CLASSIFY workspace to add phase definitions. Select in the list the phases: Biotite, Garnet, Plagioclase, Quartz; then click "Apply".
@@ -72,12 +77,12 @@ Classification is the step used to create a classified image in which each pixel
Additional information about the classification in XMapTools can be found in : Lanari, P., Tedeschi, M., (2025). Chemical map classification in XMapTools. _Applied Computing and Geosciences_, __25__, 100230 [Download].
1. Set the active workspace to CLASSIFY.
-2. Create a new training set. In the secondary tree menu, select the category "Training Set (Classification)" and press the button "Add" available in the CLASSIFY workspace to add phase definitions. Select in the list the phases: Biotite, Garnet, Plagioclase, Quartz; then click "Apply".