Added homework on pandas#5
Open
ibragimovaamina wants to merge 1 commit into
Open
Conversation
krglkvrmn
suggested changes
Dec 10, 2022
 krglkvrmn
left a comment
krglkvrmn
left a comment
There was a problem hiding this comment.
Всё круто, жалко, что без второй части
Comment on lines
+16
to
+17
| rrna_gff_df = read_gff('data/rrna_annotation.gff') | ||
| alignment_bed_df = read_bed('data/alignment.bed') |
There was a problem hiding this comment.
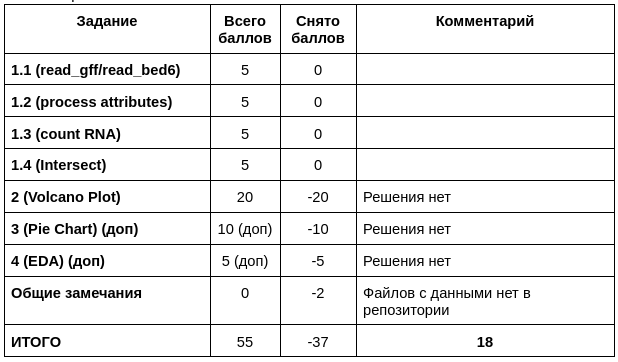
Если код подразумевает работу с данными из каких-то файлов, то эти файлы обязательно нужно класть в репозиторий и указывать относительный путь до них. Исключение только одно - слишком большие файлы, в таком случае можно заливать их маленький кусочек.
| # Function for reading gff files | ||
| def read_gff(path_to_gff): | ||
| gff_header = ['chromosome', 'source', 'type', 'start', 'end', 'score', 'strand', 'phase', 'attributes'] | ||
| return pd.read_csv(path_to_gff, sep='\t', names=gff_header, comment = '#') |
There was a problem hiding this comment.
Suggested change
| return pd.read_csv(path_to_gff, sep='\t', names=gff_header, comment = '#') | |
| return pd.read_csv(path_to_gff, sep='\t', names=gff_header, comment='#') |
| rrnas_by_types = pd.DataFrame({'count' : rrna_gff_df.groupby(['chromosome','attributes']).size()}).reset_index() | ||
|
|
||
| # Merging gff and bed files | ||
| merged_df = pd.merge(rrna_gff_df, alignment_bed_df, how='outer', left_on=['chromosome'], right_on=['chromosome']) |
There was a problem hiding this comment.
Можно чуть проще
Suggested change
| merged_df = pd.merge(rrna_gff_df, alignment_bed_df, how='outer', left_on=['chromosome'], right_on=['chromosome']) | |
| merged_df = pd.merge(rrna_gff_df, alignment_bed_df, how='outer', on='chromosome') |
| plt.xticks(rotation=90, size=10); | ||
|
|
||
| # Extracting rRNAs which intersect with alignment | ||
| rrnas_align_intersect = merged_df[(merged_df['start_x'] >= merged_df['start_y']) & (merged_df['end_x'] <= merged_df['end_y'])] |
There was a problem hiding this comment.
Рабочий вариант. Можно ещё через query, ИМХО так чуть лаконичнее
Suggested change
| rrnas_align_intersect = merged_df[(merged_df['start_x'] >= merged_df['start_y']) & (merged_df['end_x'] <= merged_df['end_y'])] | |
| rrnas_align_intersect = merged_df.query('start_x >= start_y and end_x <= end_y') |
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
Sign up for free
to join this conversation on GitHub.
Already have an account?
Sign in to comment
2 participants
Add this suggestion to a batch that can be applied as a single commit.This suggestion is invalid because no changes were made to the code.Suggestions cannot be applied while the pull request is closed.Suggestions cannot be applied while viewing a subset of changes.Only one suggestion per line can be applied in a batch.Add this suggestion to a batch that can be applied as a single commit.Applying suggestions on deleted lines is not supported.You must change the existing code in this line in order to create a valid suggestion.Outdated suggestions cannot be applied.This suggestion has been applied or marked resolved.Suggestions cannot be applied from pending reviews.Suggestions cannot be applied on multi-line comments.Suggestions cannot be applied while the pull request is queued to merge.Suggestion cannot be applied right now. Please check back later.
No description provided.